ГОСТ Р 52249-2004
Группа Р26
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРАВИЛА ПРОИЗВОДСТВА И КОНТРОЛЯ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
Good Manufacturing Practice for Medicinal Products (GMP)
ОКС 11.120.10
ОКП 94 5120
Дата введения 2005-01-01
Предисловие
Задачи, основные принципы и правила проведения работ по государственной стандартизации в Российской Федерации установлены ГОСТ Р 1.0-92 "Государственная система стандартизации Российской Федерации. Основные положения" и ГОСТ Р 1.2-92 "Государственная система стандартизации Российской Федерации. Порядок разработки государственных стандартов"
Сведения о стандарте
1 ПОДГОТОВЛЕН Ассоциацией инженеров по контролю микрозагрязнений (АСИНКОМ) по собственному аутентичному переводу, указанному в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 458 "Производство и контроль качества лекарственных средств"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Госстандарта России от 10 марта 2004 г. N 160-ст
4 Настоящий стандарт идентичен Правилам производства лекарственных средств Европейского Союза (ЕС Guide to Good Manufacturing Practice for Medicinal Products)
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в указателе "Национальные стандарты", а текст этих изменений - в информационных указателях "Национальные стандарты". В случае пересмотра или отмены настоящего стандарта соответствующая информация будет опубликована в информационном указателе "Национальные стандарты"
Введение
В мировой практике одним из важнейших документов, определяющим требования к производству и контролю качества лекарственных средств для человека и животных, являются "Правила производства лекарственных средств" - "Good Manufacturing Practice for Medicinal Products (GMP)".
Они направлены на обеспечение высокого уровня качества и безопасности лекарственных средств и гарантирование того, что лекарственное средство изготовлено в соответствии со своей формулой (составом), не содержит посторонних включений, маркировано надлежащим образом, упаковано и сохраняет свои свойства в течение всего срока годности.
Правила GMP устанавливают требования к системе управления качеством, контролю качества, персоналу, помещениям и оборудованию, документации, производству продукции и проведению анализов по контрактам, рекламациям, порядку отзыва продукции и организации самоинспекций.
Область применения
Настоящий стандарт устанавливает требования к производству и контролю качества лекарственных средств для человека и животных.
Стандарт распространяется на все виды лекарственных средств и устанавливает общие требования к их производству и контролю качества, а также специальные требования к производству отдельных видов лекарственных средств (приложения 1-18).
Стандарт не распространяется на обеспечение промышленной безопасности, пожарной безопасности, взрывобезопасности, химической безопасности и безопасности других видов при производстве лекарственных средств, требования к которым приведены в других нормативных документах.
1 Управление качеством
Принципы
1.1 Производитель лекарственных средств должен организовать их производство так, чтобы лекарственные средства гарантированно соответствовали своему назначению и предъявляемым к ним требованиям и не создавали риска для потребителей из-за нарушения условий безопасности, качества или эффективности. Ответственность за выполнение этих требований несут руководители и все работники предприятия-производителя, а также поставщики и дистрибьюторы.
Для достижения этой цели на предприятии на основе настоящего стандарта (Правил GMP) должна быть создана система обеспечения качества, включающая в себя организацию контроля качества.
Следует документально оформить в полном объеме требования к системе обеспечения качества и организовать контроль эффективности ее функционирования. Все звенья этой системы следует укомплектовать квалифицированным персоналом, обеспечить необходимыми помещениями, оборудованием и пр. Ответственность за функционирование системы возлагается, в первую очередь, на руководителей и Уполномоченных лиц.
Основные принципы обеспечения качества, Правил GMP и контроля качества взаимосвязаны и имеют первостепенное значение в организации производства лекарственных средств.
Обеспечение качества
1.2 Обеспечение качества является комплексной задачей, решение которой требует реализации всех мер, направленных на достижение заданных требований к качеству лекарственных средств. Обеспечение качества основывается на выполнении требований настоящего стандарта и других нормативных документов.
Система обеспечения качества (система качества) при производстве лекарственных средств должна гарантировать следующее:
I Лекарственные средства разработаны с учетом требований настоящего стандарта и требований к работе лабораторий.
II На все производственные и контрольные операции разработана документация в соответствии с настоящим стандартом.
Ill Ответственность и обязанности всех работников четко определены.
IV Предусмотрены меры, обеспечивающие производство, поставку и использование исходных и упаковочных материалов, соответствующих заданным требованиям.
V Контроль промежуточной продукции и технологического процесса (внутрипроизводственный контроль), аттестация (валидация) процессов и оборудования проводятся в необходимом объеме.
VI Производство и контроль готовой продукции соответствуют утвержденным инструкциям (методикам).
VII Реализация лекарственных средств до выдачи Уполномоченным лицом разрешения на выпуск исключена. Уполномоченное лицо должно подтвердить, что каждая серия продукции произведена и проверена в соответствии с установленными требованиями.
VIII Существующая система мер обеспечивает уровень качества лекарственных средств при их хранении, отгрузке и последующем обращении в течение всего срока годности.
IX Порядок проведения самоинспекции и/или аудита качества позволяет регулярно оценивать эффективность системы обеспечения качества.
Требования к производству и контролю качества лекарственных средств
1.3 Настоящий стандарт входит в систему качества и направлен на обеспечение гарантии того, что производство и контроль качества продукции постоянно соответствуют требованиям, установленным в документации.
Основные требования
I Все производственные процессы должны быть четко регламентированы и периодически пересматриваться с учетом накопленного опыта. Следует контролировать стабильность производства лекарственных средств с заданным качеством в соответствии со спецификациями на них.
II Следует проводить аттестацию (валидацию) критических стадий процессов производства, в том числе при внесении существенных изменений в технологический процесс.
Ill Следует обеспечить все необходимые условия для выполнения требований настоящего стандарта, в т.ч. включая наличие:
a) обученного и аттестованного персонала;
b) необходимых помещений и площадей;
c) соответствующего оборудования и системы обслуживания;
d) материалов, средств упаковки и маркировки, удовлетворяющих заданным требованиям;
e) утвержденных инструкций и методик;
f) требуемых условий хранения и транспортирования.
IV Инструкции и методики должны быть конкретными, изложены ясно и однозначно в письменной форме.
V Персонал должен быть обучен правильному выполнению инструкций.
VI В процессе производства следует составлять протоколы (заполняемые в рукописной форме и/или с использованием технических средств), документально подтверждающие фактическое проведение предусмотренных инструкциями технологических стадий и получение продукции требуемого качества в количестве, соответствующем установленным нормам. Все отклонения необходимо расследовать и протоколировать в полном объеме.
VII Протоколы на серию, в т.ч. документацию по реализации продукции, должны давать возможность прослеживать изготовление каждой серии продукции и должны храниться в полном объеме в доступной форме.
VIII Порядок реализации (оптовой продажи) продукции должен сводить к минимуму любой риск для ее качества.
IX Следует организовать систему отзыва любой серии продукции из продажи или поставки.
X Рекламации на качество продукции следует тщательно рассматривать, а причины ухудшения качества расследовать с принятием соответствующих мер по их предотвращению.
Контроль качества
1.4 Контроль качества включает в себя отбор проб, проведение испытаний (анализов) и оформление соответствующей документации. Инструкции по организации, документированию и выдаче разрешения на выпуск продукции должны включать в себя проведение всех необходимых испытаний и запрещать использование исходного сырья и материалов и реализацию готовой продукции до подтверждения соответствия качества установленным требованиям.
Основные требования к контролю качества
I Наличие необходимых помещений и оборудования, обученного персонала, утвержденных методик по отбору проб, проверке и проведению испытаний исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции, контролю окружающей среды, в случае необходимости.
II Проведение отбора проб исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции аттестованным персоналом в соответствии с методиками, утвержденными отделом контроля качества.
Ill Проведение испытаний аттестованными (валидированными) методами.
IV Составление протоколов (заполняемых рукописным способом и/или с применением технических средств), подтверждающих фактическое проведение всех необходимых отборов проб, проверок и испытаний, а также регистрацию любых отклонений и расследований в полном объеме.
V Подтверждение того, что готовая продукция содержит активные ингредиенты по качественному и количественному составу, соответствующие регистрационному досье, имеет требуемую чистоту, правильно упакована и маркирована.
VI Оформление протоколов проверки исходного сырья и материалов, промежуточной, нерасфасованной и готовой продукции, их анализ и сравнение со спецификациями. Оценка продукции включает в себя изучение всей необходимой производственной документации и анализ отклонений от установленных требований.
VII Получение разрешения на продажу или поставку любой серии продукции только после подтверждения Уполномоченным лицом ее соответствия регистрационному досье.
VIII Сохранение достаточного количества образцов исходных материалов и продукции для возможной проверки в случае необходимости. Образцы продукции следует хранить в своей окончательной упаковке, за исключением крупных упаковок.
2 Персонал
Принципы
Организация и функционирование производства и системы обеспечения качества лекарственных средств зависят от персонала. Предприятие должно быть укомплектовано персоналом необходимой численности и квалификации. Должностные обязанности каждого сотрудника должны быть оформлены документально и усвоены каждым сотрудником. Все сотрудники также должны знать требования настоящего стандарта (Правил GMP), относящиеся к сфере их деятельности, и проходить начальное и повторное обучение в необходимом объеме, в т.ч. по правилам личной гигиены.
Общие положения
2.1 Персонал, работающий в производстве лекарственных средств, должен обладать необходимой квалификацией и практическим опытом. Должностные обязанности отдельного сотрудника не должны быть слишком объемными и способствующими его чрезмерной загруженности, отрицательно влияющей на качество продукции.
2.2 На предприятии должна быть четкая организационная структура. Служебные обязанности руководящих работников должны быть изложены в должностных инструкциях. Руководители должны обладать достаточными полномочиями для выполнения своих функций. Их полномочия могут быть переданы официально назначенным заместителям, имеющим достаточную квалификацию. Следует исключить дублирование ответственности сотрудников, связанной с выполнением требований настоящего стандарта.
Руководящие работники
2.3 Руководители производства, руководитель службы (отдела) контроля качества и Уполномоченное лицо (лица) должны быть заняты на предприятии полный рабочий день. Руководители производства и службы (отдела) контроля качества должны быть независимыми друг от друга. На больших предприятиях часть функций, перечисленных в 2.5-2.7, передается, при необходимости, другим сотрудникам.
2.4 Обязанности Уполномоченных лиц
a) Для лекарственных средств, выпущенных в Российской Федерации, Уполномоченное лицо должно гарантировать, что каждая серия продукции была произведена и проверена в соответствии с установленными требованиями;
b) Для лекарственных средств, выпущенных вне Российской Федерации, Уполномоченное лицо должно гарантировать, что импортируемая серия продукции прошла проверку в порядке, установленном для России;
c) До выдачи разрешения на выпуск лекарственных средств в сферу обращения Уполномоченное лицо должно документально подтвердить, что каждая серия продукции удовлетворяет соответствующим документам.
Квалификация Уполномоченного лица должна соответствовать установленным требованиям. Уполномоченное лицо должно входить в штат предприятия - производителя лекарственных средств. Его обязанности могут быть переданы только лицам, имеющим статус Уполномоченного лица.
2.5 Основные обязанности руководителя производства
I Организация производства и хранения продукции в соответствии с документацией с целью обеспечения требуемого качества.
II Утверждение инструкций, связанных с производственным процессом, и обеспечение их точного выполнения.
Ill Контроль за рассмотрением и подписанием всех производственных протоколов лицами, имеющими необходимые полномочия, до передачи их в службу контроля качества.
IV Контроль за работой своего подразделения, содержанием помещений, эксплуатацией и техническим обслуживанием оборудования.
V Контроль за проведением работ по аттестации (валидации).
VI Организация первичного и последующего обучения производственного персонала.
2.6 Основные обязанности руководителя службы контроля качества
I Утверждение или отклонение исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции.
II Оценка протоколов на серию продукции.
Ill Проведение необходимых испытаний.
IV Утверждение спецификаций, инструкций по отбору проб, методик испытаний и других методик по контролю качества.
V Допуск к работе специалистов-аналитиков, работающих по контракту, и контроль за их деятельностью.
VI Контроль работы подведомственного отдела, обслуживания его помещений и оборудования.
VII Контроль проведения аттестации (валидации).
VIII Организация первичного и последующего обучения персонала подведомственного отдела.
Другие обязанности сотрудников отдела контроля качества приведены в разделе 6.
2.7 Руководители производства и отдела контроля качества имеют ряд совместных обязанностей, относящихся к обеспечению качества продукции. Эти обязанности с учетом действующих норм и правил могут включать в себя следующее:
- утверждение письменных инструкций, методик и других документов, в т.ч. внесение изменений в них;
- контроль за окружающей средой на производстве;
- контроль за соблюдением правил производственной гигиены;
- аттестацию (валидацию) процессов;
- обучение персонала;
- утверждение и контроль за поставщиками исходных материалов;
- утверждение и контроль за производителями, работающими по контракту;
- определение условий хранения материалов, продукции и контроль за их соблюдением;
- хранение протоколов;
- постоянный контроль соответствия требованиям настоящего стандарта;
- проведение инспекций, расследований и отборов проб с целью выявления факторов, способных повлиять на качество продукции.
Обучение
2.8 Предприятие-производитель должно обеспечить обучение всех сотрудников, занятых производством или контролем качества (в т.ч. технический, обслуживающий персонал и персонал, выполняющий уборку), а также других сотрудников, деятельность которых может повлиять на качество продукции.
2.9 Помимо базового обучения требованиям настоящего стандарта (по теории и практике GMP) вновь принятые сотрудники должны пройти обучение в соответствии с их должностными обязанностями. Следует организовать периодическое обучение персонала и оценивать эффективность этого обучения на практике. Обучение следует проводить по программам, утвержденным руководителями производства или службы (отдела) контроля качества. Протоколы обучения должны храниться на предприятии.
2.10 Сотрудники, работающие в зонах, в которых загрязнение представляет опасность, например, в чистых зонах или в зонах работы с сильнодействующими, токсичными, инфицирующими или сенсибилизирующими веществами, должны пройти специальное обучение.
2.11 Посетители и/или необученные сотрудники не должны допускаться в зоны, связанные с производством и контролем качества. При необходимости они должны предварительно пройти инструктаж по правилам личной гигиены, порядку переодевания и ношению специальной одежды. За этими лицами должен быть организован тщательный контроль.
2.12 При обучении следует подробно разъяснять и обсуждать концепцию обеспечения качества для ее полного усвоения и дальнейшего применения.
Гигиена персонала
2.13 На предприятии должны быть разработаны правила личной гигиены персонала с учетом особенностей конкретного производства. Правила должны содержать инструкции, регламентирующие требования к состоянию здоровья, соблюдению гигиены и порядку ношения одежды. Инструкции должны соблюдать все сотрудники, которые связаны с нахождением в производственных помещениях и помещениях контроля качества. Руководство предприятия несет ответственность за выполнение персоналом правил гигиены и организацию необходимого обучения.
2.14 Все лица, принимаемые на работу, должны проходить медицинский осмотр. На предприятии должны быть инструкции с перечнем показателей состояния здоровья, которые могут оказать влияние на качество продукции. В случаях, связанных с производственной необходимостью или состоянием здоровья, сотрудники должны проходить повторный медицинский осмотр.
2.15 Лица с инфекционными заболеваниями и повреждениями на открытых участках тела не допускаются к производству лекарственных средств.
2.16 Одежда входящего в производственные помещения должна соответствовать назначению этого помещения.
2.17 В производственных и складских зонах запрещаются курение, прием пищи или питье, жевание резинки, а также хранение пищевых продуктов, напитков, табачных изделий и личных лекарственных средств. Не допускается любая деятельность, нарушающая правила гигиены в производственных помещениях или других местах, которая может оказать отрицательное влияние на качество продукции.
2.18 Непосредственный контакт операторов с открытой продукцией или любыми деталями оборудования, контактирующими с продукцией, не допускается.
2.19 Персонал должен пройти инструктаж по правилам мытья рук.
2.20 Специальные требования, относящиеся к производству отдельных видов продукции, например, стерильных препаратов, даны в приложениях к настоящему стандарту.
3 Помещения и оборудование
Принципы
Место расположения, проект, строительство, монтаж, оснащение и обслуживание помещений и оборудования должны соответствовать характеру выполняемых работ. Планировка помещений и конструкция оборудования должны минимизировать риск ошибок, предусматривать проведение эффективной уборки и обслуживания с целью предотвращения перекрестного загрязнения, появления пыли или грязи и, в общем случае, устранения любого фактора, ухудшающего качество продукции.
Помещения
Общие положения
3.1 Риск загрязнения материалов и продукции, создаваемый окружающей средой производственных помещений (зданий), должен быть минимальным при условии соблюдения всех мер защиты.
3.2 При эксплуатации помещений следует выполнять меры предосторожности, при этом проведение технического обслуживания и ремонта не должно оказывать вредного влияния на качество продукции. Уборка и дезинфекция помещений должны выполняться в соответствии с письменными инструкциями.
3.3 Освещение, температурный режим, влажность и вентиляция должны соответствовать назначению помещения и не оказывать прямого или косвенного отрицательного влияния на работу оборудования и лекарственные средства во время их изготовления и хранения.
3.4 При проектировании и эксплуатации помещений следует предусмотреть максимальную защиту от проникания в них насекомых или животных.
3.5 В помещения не допускаются лица, не имеющие права доступа в них. Производственные, складские помещения и помещения контроля качества не должны использоваться для сквозного прохода персонала, не работающего в них.
Производственная зона
3.6 Для минимизации риска для здоровья людей из-за перекрестных загрязнений при производстве некоторых лекарственных средств, таких как сенсибилизирующие вещества (например, пенициллины) или биологические препараты (например, из живых микроорганизмов), следует предусмотреть специальные и изолированные технические средства (помещения, оборудование, средства обслуживания и др.). В одних и тех же помещениях не допускается производство отдельных видов антибиотиков, некоторых гормонов, цитотоксинов, сильнодействующих лекарственных средств и продукции немедицинского назначения. В исключительных случаях производство таких препаратов допускается в одних помещениях при разделении циклов производства по времени, с соблюдением специальных мер предосторожности и проведением необходимой аттестации (валидации). В зданиях, используемых для производства лекарственных средств, не допускается производство ядов технического назначения (пестицидов и гербицидов).
3.7 Планировочные решения помещений по возможности должны соответствовать логической последовательности производственных операций и обеспечивать выполнение требований к чистоте.
3.8 Планировочные решения рабочих зон и зон хранения внутри производства должны обеспечивать последовательное и логичное размещение оборудования и материалов, сводить к минимуму риск перепутывания различных лекарственных средств или их компонентов, перекрестного загрязнения и ошибочного выполнения или пропуска любых операций по производству или контролю.
3.9 Если исходные и первичные упаковочные материалы, промежуточные или нерасфасованные продукты подвергаются воздействию окружающей среды, внутренние поверхности помещений (стены, пол и потолок) должны быть гладкими, не иметь открытых соединений и трещин, не выделять частиц и должны обеспечивать возможность беспрепятственной и эффективной уборки и дезинфекции.
3.10 Конструкция и размещение труб, осветительных приборов, оборудования вентиляции и т.п. не должны иметь мест, труднодоступных для очистки. По возможности их обслуживание должно осуществляться с внешней стороны производственных помещений.
3.11 Трубопроводы для стоков (канализация) должны иметь необходимые размеры и быть оборудованы устройствами, предотвращающими обратный поток. Следует избегать открытых желобов. При необходимости они должны быть неглубокими для удобства очистки и дезинфекции.
3.12 В производственных зонах, в зависимости от выпускаемой продукции, выполняемых операций и требований к окружающей среде, следует предусматривать эффективную систему вентиляции с обеспечением требуемой температуры и, при необходимости, влажности и очистки воздуха.
3.13 Исходные материалы взвешивают, как правило, в специально оборудованных для этого помещениях.
3.14 Если выполнение работы сопровождается выделением пыли (например, при отборе проб, взвешивании, смешении, производственных операциях и упаковке сухих продуктов), то необходимо предусмотреть меры по предотвращению перекрестного загрязнения и проведению очистки.
3.15 При проектировании (в т.ч. разработке планировочных решений) помещений для упаковки лекарственных средств следует предусматривать специальные меры против перепутывания или перекрестного загрязнения материалов и продукции.
3.16 Производственные помещения должны быть хорошо освещены, особенно в местах выполнения визуального контроля.
3.17 Внутрипроизводственный контроль может проводиться в зоне производства, если это не создает помех для технологического процесса.
Зоны складирования
3.18 Зоны складирования должны быть достаточной вместимости для обеспечения надлежащего хранения различных категорий материалов и продукции (исходного сырья и упаковочных материалов; промежуточной, нерасфасованной и готовой продукции; продукции, находящейся в карантине, разрешенной для выпуска, отклоненной, возвращенной или отозванной продукции).
3.19 При проектировании и организации зон складирования следует предусматривать надлежащие условия хранения. Зоны складирования должны быть чистыми, сухими и иметь требуемый температурный режим. При необходимости следует обеспечивать специальные условия хранения (температуру, влажность и т.п.) и их контроль.
3.20 В зонах приемки и выдачи материалов и продукции должна быть обеспечена их защита от неблагоприятных погодных условий. Проект зоны приемки должен предусматривать очистку упаковок с поступающими материалами перед их складированием.
3.21 Если режим карантина обеспечивается хранением продукции в раздельных зонах, то эти зоны должны иметь четкое обозначение. Доступ в них должен быть разрешен только лицам, имеющим на это право. Любая другая система, заменяющая физическое разделение, должна обеспечивать эквивалентную безопасность.
3.22 Отбор проб исходных материалов, как правило, следует выполнять в отдельной зоне. При отборе проб в складской зоне необходимо принять меры против прямого или перекрестного загрязнения.
3.23 Отклоненные, отозванные или возвращенные материалы и продукцию следует хранить в изолированных зонах.
3.24 Сильнодействующие вещества и препараты должны храниться в безопасных и охраняемых помещениях.
3.25 Следует обеспечить надежное и безопасное хранение печатных материалов ввиду их ключевой роли в подтверждении идентичности лекарственных средств.
Зоны контроля качества
3.26 Как правило, лаборатории контроля качества должны быть отделены от производственных помещений. Это особенно важно для лабораторий контроля биологических, микробиологических препаратов или радиоизотопов, которые также должны быть отделены друг от друга.
3.27 Проект контрольных лабораторий должен соответствовать требованиям к выполняемым в них операциям. Площадь лабораторий должна быть достаточной для исключения перепутывания и перекрестного загрязнения, а также для хранения образцов и протоколов.
3.28 Для размещения чувствительных приборов, нуждающихся в защите от электромагнитных полей, вибрации, повышенной влажности или других внешних факторов, могут быть предусмотрены отдельные помещения.
3.29 Особые требования предъявляются к лабораториям, в которых проводятся работы с образцами специфических веществ, например, биологическими или радиоактивными материалами.
Вспомогательные зоны
3.30 Комнаты отдыха и приема пищи должны быть отделены от производственных помещений.
3.31 Помещения для переодевания и хранения одежды, туалеты и душевые должны иметь удобный доступ, их планировка и размеры должны соответствовать численности персонала. Не допускается выход из туалетов непосредственно в производственные или складские зоны.
3.32 Ремонтные участки должны быть, по возможности, отделены от производственных помещений. При необходимости хранения запасных частей и инструментов в зоне производства должны быть предусмотрены специальные помещения или шкафы.
3.33 Помещения для содержания животных должны быть изолированы от остальных зон, оборудованы отдельными системами подготовки воздуха и отдельным входом.
Оборудование
3.34 Конструкция, монтаж и порядок технического обслуживания оборудования должны соответствовать его назначению.
3.35 Работы по ремонту и техническому обслуживанию оборудования не должны оказывать отрицательного влияния на качество продукции.
3.36 Конструкция производственного оборудования должна обеспечивать удобство и возможность его очистки. Операции по очистке оборудования должны выполняться в соответствии с подробными письменными инструкциями. Оборудование следует содержать в сухом и чистом состоянии.
3.37 Инвентарь и материалы для очистки не должны быть источниками загрязнения.
3.38 Оборудование должно быть установлено так, чтобы, по возможности, исключить риск загрязнения или выполнения ошибочных действий.
3.39 Технологическое оборудование не должно влиять на качество продукции и представлять какую-либо опасность. Части технологического оборудования, контактирующие с продукцией, не должны вступать с ней в реакцию, выделять или абсорбировать вещества, оказывающие влияние на качество продукции, в такой степени, чтобы это могло представлять опасность.
3.40 Погрешность приборов для измерения массы и другого измерительного оборудования должна соответствовать производственным и контрольным операциям, в которых они используются.
3.41 Периодичность калибровки (поверки) измерительных, регистрирующих, контрольных приборов и оборудования для измерения массы должна соответствовать указанной в соответствующих методиках. Результаты калибровки (поверки) должны быть документированы.
3.42 Стационарные трубопроводы должны быть маркированы с указанием проходящих по ним веществ и, если требуется, направления потока.
3.43 Системы трубопроводов для воды очищенной и воды для инъекций (дистиллированной, деионизованной и др.) следует обрабатывать в соответствии с инструкциями, в которых указаны уровни действия по микробному загрязнению и требуемые корректирующие меры.
3.44 Неисправное оборудование должно быть изъято из зоны производства и контроля качества или обозначено соответствующим образом.
4 Документация
Принципы
Правильно составленная документация является важной частью системы обеспечения качества. Четкое оформление документации позволяет предотвратить ошибки, возможные при устном общении, и проследить все этапы производства конкретной серии продукции. Спецификации, промышленные регламенты, инструкции, методики и протоколы серии продукции должны быть оформлены надлежащим образом и не должны содержать ошибок.
Общие положения
4.1 Виды документов
спецификация (specification): Документ, содержащий требования к материалам и продуктам, используемым или получаемым при производстве, являющийся основой для оценки качества лекарственных средств.
промышленный регламент, технологическая инструкция и инструкция по упаковке (manufacturing formulae, processing and packaging instructions): Документы, определяющие все используемые исходные материалы и операции по производству и упаковке продукции.
инструкция, методика, процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования).
протокол на серию (record): Документ, отражающий ход производства каждой серии продукции, в т.ч. разрешение на ее реализацию, и все факторы, влияющие на качество готовой продукции.
4.2 Следует ввести четкую систему разработки, оформления, распространения и пересмотра документов. Должен быть установлен порядок их выдачи, внесения изменений и изъятия. Документы должны соответствовать регистрационному досье.
4.3 Документы должны быть подписаны и утверждены лицами, имеющими право подписи, с указанием даты.
4.4 Документ не должен допускать двусмысленного толкования. Название, вид и назначение документа должны быть четкими и ясными. Документ должен иметь логичную структуру, обеспечивающую простоту его проверки. Копии с документов должны быть ясными и четкими. Способ снятия копий с рабочих документов должен исключать возможность появления ошибок.
4.5 Документы следует регулярно пересматривать и актуализировать. При пересмотре документа необходимо исключить использование устаревшей версии.
4.6 Не допускается оформление документов в рукописном виде. При необходимости данные в документ вносят четким, разборчивым почерком так, чтобы внесенные данные нельзя было удалить. Для внесения данных в документе должно быть предусмотрено достаточно свободного места.
4.7 При внесении изменений в документы следует проставлять дату внесения изменения и подпись лица, сделавшего это изменение. При необходимости следует указать причину внесения изменений. Внесенные изменения не должны препятствовать восприятию исходного текста.
4.8 Протоколы следует оформлять одновременно с выполнением соответствующих действий таким образом, чтобы можно было проследить все основные операции при производстве лекарственных средств. Протоколы следует хранить не менее одного года со дня окончания срока годности готовой продукции.
4.9 Запись данных может выполняться с использованием электронной техники, фотографирования или других средств, обеспечивающих надежное хранение информации в соответствии с инструкциями по использованию этих средств. Следует проверять точность записей. При ведении документации в электронном виде право доступа или изменения данных в компьютере могут иметь только лица с соответствующими полномочиями, при этом следует вести протокол изменений и изъятий. Для ограничения доступа к электронной базе данных следует использовать систему паролей или других средств; внесение особо важных данных должно проверяться независимым способом. При хранении протоколов на серии продукции в электронном виде для защиты от потери информации необходимо создавать резервные копии на магнитных носителях, микрофильмах, бумаге или иных надежных средствах. В период хранения эти данные должны быть доступными.
Обязательная документация
Спецификации
4.10 Спецификации составляются и утверждаются на исходные, упаковочные материалы и готовую продукцию. При необходимости составляются спецификации на промежуточную и нерасфасованную продукцию.
Спецификации на исходные и упаковочные материалы
4.11 Спецификации на исходные материалы, первичную упаковку или печатные материалы должны включать в себя следующие данные:
a) описание материалов, в т.ч.:
- наименование и внутризаводской код;
- ссылку на фармакопейную статью (при ее наличии);
- наименование утвержденных поставщиков и, по возможности, первичного производителя материалов;
- образец печатных материалов;
b) методики отбора проб и проведения испытаний или ссылки на соответствующие методики;
c) количественные и качественные характеристики с указанием допустимых пределов;
d) условия хранения и меры предосторожности;
e) максимальный срок хранения до повторного контроля.
Спецификации на промежуточную и нерасфасованную продукцию
4.12 Спецификации на промежуточную и нерасфасованную продукцию составляются при поступлении ее на предприятие, реализации или использовании промежуточной продукции при оценке качества готовой продукции. Эти спецификации должны быть аналогичны спецификациям на исходные материалы или готовую продукцию в зависимости от характера использования промежуточной или нерасфасованной продукции.
Спецификации на готовую продукцию
4.13 Спецификации на готовую продукцию должны включать в себя следующие данные:
a) наименование продукции и код (при необходимости);
b) состав препарата или ссылку на соответствующий документ;
c) описание лекарственной формы и данные об упаковке;
d) методики отбора проб и проведения испытаний (или ссылки на них);
e) количественные и качественные характеристики с указанием допустимых пределов;
f) условия хранения и особые меры предосторожности при обращении с препаратом (при необходимости);
g) срок годности.
Промышленный регламент и технологические инструкции
Для каждого вида продукции и размера серии продукции должен быть разработан и утвержден промышленный регламент.
Промышленный регламент включает в себя данные о продукте и технологические инструкции.
Промышленный регламент может содержать инструкции по упаковке и другие разделы.
4.14 Данные о продукте включают в себя:
a) наименование и код в соответствии со спецификацией;
b) описание лекарственной формы, ее дозировки и размер серии;
c) перечень всех исходных материалов с точным наименованием в соответствии с принятой номенклатурой и указанием их кодов, а также указанием всех веществ, которые могут преобразовываться в ходе технологического процесса;
d) ожидаемый выход готовой продукции с указанием допустимых пределов и выход промежуточных продуктов (при необходимости).
4.15 Технологические инструкции включают в себя:
a) данные о месте нахождения производства и основном оборудовании;
b) инструкции по подготовке основного оборудования (например, по очистке, сборке, калибровке (поверке), стерилизации) или ссылки на них;
c) подробное постадийное описание технологического процесса (например, по контролю материалов, предварительной обработке, последовательности внесения материалов, времени перемешивания, температуре и т.д.);
d) описание всех видов внутрипроизводственного контроля с указанием допустимых пределов;
e) условия хранения нерасфасованной продукции (в т.ч. требования к упаковке, маркировке) и специальные условия хранения (при необходимости);
f) специальные меры предосторожности.
Инструкции по упаковке
4.16 Для каждого вида продукции, размера и вида упаковки должны быть разработаны и утверждены инструкции по упаковке, которые должны включать в себя:
a) наименование продукции;
b) описание ее лекарственной формы и дозировки (при необходимости);
c) количество продукта в окончательной упаковке, выраженное в единицах измерения (штуках, массе или объеме);
d) перечень всех упаковочных материалов, необходимых для серии продукции стандартного размера, в т.ч. количество, размер и тип упаковочного материала с указанием кода или номера в соответствии с их спецификацией;
e) образец или копию соответствующего печатного упаковочного материала (при необходимости) и образцы с указанием места нанесения номера серии и срока годности продукта;
f) специальные меры предосторожности, в т.ч. тщательную проверку оборудования и зоны упаковки, гарантирующие чистоту упаковочной линии перед началом работы;
g) описание процесса упаковки со всеми основными вспомогательными операциями и используемым оборудованием;
h) подробное описание проведения внутрипроизводственного контроля, в т.ч. порядок отбора проб и указание допустимых пределов.
Дополнительно могут быть разработаны другие документы, конкретизирующие положения промышленного регламента.
Протоколы на серию продукции
4.17 На каждую серию продукции составляется протокол (протокол на серию продукции). Протоколы хранятся в установленном порядке. Протоколы должны быть составлены в соответствии с промышленными регламентами и технологическими инструкциями. Методика составления протоколов должна исключать ошибки при их заполнении. В протоколе должен быть указан номер произведенной серии продукции.
Перед началом любого технологического процесса необходимо проверить и оформить протокол о том, что оборудование и рабочее место находятся в чистом состоянии, не содержат остатков предыдущего продукта, документации и материалов, не относящихся к данному процессу, и готово к использованию.
Протокол на серию продукции должен включать в себя следующие данные:
а) наименование продукта;
b) дату и время начала и окончания основных промежуточных этапов и полного технологического процесса;
c) фамилию и инициалы сотрудника, ответственного за выполнение каждой производственной стадии;
d) фамилию(и) и инициалы оператора(ов), ответственного(ых) за основные стадии производства, а также фамилии и инициалы лиц, проверявших выполнение каждой из этих операций (например, взвешивания);
e) номер серии и/или номер анализа, а также фактическое количество взвешенных исходных материалов, в т.ч. номер серии и количество добавленных регенерированных или переработанных материалов;
f) основные технологические операции или действия, а также основное оборудование;
g) протоколы внутрипроизводственного контроля с указанием исполнителей и полученных результатов;
h) выход продукции, полученной на основных производственных стадиях;
i) подробное описание любых отклонений от промышленного регламента и технологических инструкций, подписанное ответственным лицом.
Эта информация оформляется документально одновременно с выполнением соответствующей операции. Протокол на серию продукции подписывается лицом, ответственным за проведенный технологический процесс, с указанием даты.
Протоколы на упаковку серии продукции
4.18 Для каждой серии или части серии продукции должны составляться протоколы на упаковку, которые хранятся в установленном порядке. Протоколы должны быть основаны на действующих инструкциях по упаковке. Методика составления протоколов должна исключать ошибки при заполнении. В протоколах указываются номер серии и количество нерасфасованной продукции, подлежащей упаковке, а также номер серии и планируемое количество готовой продукции.
Перед началом любой упаковочной операции необходимо проверить и оформить протокол о том, что оборудование и рабочее место находятся в чистом состоянии, не содержат остатков предыдущего продукта, документации и материалов, не относящихся к данному процессу, и готовы к использованию.
Протоколы на упаковку серии продукции должны включать в себя следующие данные:
a) наименование продукта;
b) дату(ы) и время операций по упаковке;
c) фамилии и инициалы лица, ответственного за упаковку;
d) фамилии и инициалы упаковщиков для различных стадий упаковки;
e) протоколы проверки соответствия упаковки требованиям инструкций по упаковке, в т.ч. результаты внутрипроизводственного контроля;
f) подробные данные о выполнении операций по упаковке, в т.ч. ссылки на используемое оборудование и упаковочные линии;
g) образцы печатных материалов, в т.ч. образцы с обозначением номера серии, срока годности и любой дополнительной информации;
h) подробное описание любых отклонений от инструкций по упаковке, подписанное ответственным лицом;
i) количество и наименование выданных, использованных, уничтоженных или возвращенных на склад печатных материалов и нерасфасованной продукции и количество полученной готовой продукции для составления общего баланса.
Эти данные оформляются документально одновременно с выполнением соответствующей операции. Протокол на упаковку серии продукции подписывается лицом, ответственным за упаковку, с указанием даты.
Инструкции и протоколы
Приемка
4.19 Приемка каждой серии каждого вида поставляемых исходных, упаковочных и печатных материалов ведется в соответствии с письменной инструкцией. По результатам приемки составляется протокол.
4.20 Протоколы приемки должны включать в себя следующие данные:
a) наименование материала по накладной и обозначению на упаковке;
b) внутризаводское наименование или код материала (при отличии от перечисления а);
c) дату приемки;
d) наименование поставщика и производителя (по возможности);
e) номер серии производителя;
f) общее количество полученных материалов и число единиц упаковки;
g) номер, присвоенный после приемки;
h) замечания (например, о состоянии упаковки).
4.21 Следует разработать и утвердить инструкции по внутризаводской маркировке, карантину и хранению исходных, упаковочных и других материалов.
Отбор проб
4.22 В инструкциях по отбору проб должны быть указаны лица, уполномоченные на проведение этих операций, используемые методики и оборудование, количество отбираемых материалов и меры предосторожности, исключающие загрязнение или ухудшение качества продукции (6.13).
Проведение испытаний
4.23 Следует разработать и утвердить методики испытаний материалов и продукции на различных этапах производства с указанием используемых методов и оборудования. Результаты испытаний оформляются в виде протокола (6.17).
Прочее
4.24 Следует разработать и утвердить инструкции, регламентирующие выдачу разрешений на реализацию и отклонение материалов и продукции, в частности, разрешений на реализацию готовой продукции Уполномоченными лицами (2.4).
4.25 Протоколы реализации каждой серии продукции следует хранить для обеспечения (при необходимости) возможности ее оперативного отзыва (раздел 8).
4.26 Следует разработать и утвердить методики (инструкции) на следующие работы:
- аттестацию (валидацию);
- монтаж и калибровку (поверку) оборудования;
- техническое обслуживание, очистку и дезинфекцию;
- обучение персонала, обеспечение его одеждой, соблюдение правил личной гигиены и пр.;
- контроль окружающей среды;
- борьбу с паразитами, насекомыми и другими животными;
- рекламации;
- отзыв продукции;
- возврат продукции.
Выполнение каждой из указанных работ оформляется протоколом.
4.27 Следует разработать и утвердить инструкции по эксплуатации основного производственного и контрольно-аналитического оборудования.
4.28 Для критического оборудования следует вести журналы с перечислением в них всех операций по аттестации (валидации), калибровке (поверке), обслуживанию, очистке или ремонту, в т.ч. дат, фамилий и инициалов, а также подписей лиц, проводивших эти операции.
4.29 Использование основного или критического оборудования и производственных площадей, где выполнялись технологические операции, следует регистрировать в специальных журналах в хронологическом порядке.
5 Производство
Принципы
Для получения продукции требуемого качества технологические операции следует выполнять согласно промышленному регламенту и соответствующим инструкциям, требованиям настоящего стандарта, нормативных документов и регистрационного досье.
Общие положения
5.1 Производственный процесс и его контроль должны выполняться квалифицированным персоналом.
5.2 Все операции с материалами и продукцией (например, приемка, карантин, отбор проб, хранение, маркировка, подготовка, приготовление, упаковка и отгрузка) должны выполняться согласно письменным инструкциям или методикам и, при необходимости, протоколироваться.
5.3 Все поступающие материалы должны быть проверены на соответствие заказу. Тару и упаковку следует очищать и маркировать.
5.4 Факты повреждения тары и упаковки, которые могут оказать отрицательное влияние на качество материалов, следует расследовать и протоколировать с последующим сообщением в отдел контроля качества.
5.5 Поступающие материалы и произведенная готовая продукция должны немедленно помещаться в карантин, действующий по принципу раздельного хранения или за счет организационных мер, и содержаться в нем до получения разрешения на использование или отгрузку.
5.6 Приемка промежуточной и нерасфасованной продукции выполняется по правилам, действующим для исходных материалов.
5.7 Все материалы и продукцию следует хранить в соответствующих условиях, определяемых производителем, в порядке, обеспечивающем разделение серий продукции и ее оборот на складе.
5.8 Для гарантии отсутствия отклонений за допустимые пределы следует обеспечить контроль выхода продукции и количественное сопоставление его с данными промышленного регламента.
5.9 Не допускается одновременное или последовательное проведение операций с различными продуктами в одном и том же помещении при отсутствии защиты от риска перепутывания или перекрестного загрязнения.
5.10 Продукция и материалы должны быть защищены от микробного и других видов загрязнений на всех этапах производства.
5.11 При работе с сухими материалами и продуктами необходимо принимать особые меры предосторожности по предотвращению образования и распространения пыли, особенно при работе с сильнодействующими и сенсибилизирующими веществами.
5.12 В ходе выполнения технологического процесса на всех материалах, упаковках с нерасфасованной продукцией, основном оборудовании и помещениях должны быть обозначения (маркировка) с указанием производимой продукции или материала, его дозировкой (при необходимости) и номера серии. При необходимости следует указывать стадию технологического процесса.
5.13 Обозначения (маркировка) на упаковке, оборудовании или помещениях должны быть четкими, однозначными, установленной формы. Кроме применения буквенных обозначений рекомендуется использовать цветовую маркировку, указывающую статус продукции (например, "Карантин", "Принято", "Отклонено", "Чистое" и т.п.).
5.14 Следует контролировать правильность соединения трубопроводов и другого оборудования, служащего для транспортирования продукции из одной зоны в другую.
5.15 Не допускается отклонение от инструкций. При необходимости письменное разрешение на отклонение от инструкций должно быть получено от компетентных лиц и отдела контроля качества.
5.16 В производственные помещения может входить только персонал, имеющий право доступа в них.
5.17 Как правило, в помещениях и на оборудовании, предназначенных для производства лекарственных средств, не допускается изготовление продукции немедицинского назначения.
Предотвращение перекрестного загрязнения при производстве
5.18 Следует исключить возможность загрязнения исходных материалов или продуктов другими материалами или продуктами. В процессе производства риск случайного перекрестного загрязнения возникает при неконтролируемом выделении пыли, газов, испарений, аэрозолей или микроорганизмов из материалов (продукции) и от остаточных загрязнений на оборудовании и одежде людей. Степень риска зависит от типа загрязнения и продукта, подверженного загрязнению.
К наиболее опасным загрязняющим веществам относятся сенсибилизирующие вещества, биологические препараты, содержащие живые микроорганизмы, некоторые гормоны, цитотоксины и другие сильнодействующие вещества. Особенно опасно загрязнение инъекционных препаратов, а также препаратов, предназначенных для приема в больших дозах и/или в течение продолжительного времени.
5.19 Для предотвращения перекрестного загрязнения следует предусмотреть следующие технические и организационные меры:
a) производство в выделенных зонах (обязательное для пенициллинов, живых вакцин, бактериальных препаратов из живых микроорганизмов и некоторых других биологических препаратов) или разделение циклов производства во времени с соответствующей уборкой помещения и оборудования между циклами;
b) организация воздушных шлюзов и вытяжных устройств;
c) снижение риска загрязнения, вызываемого рециркуляцией или повторным поступлением необработанного или недостаточно обработанного воздуха;
d) хранение защитной (специальной) одежды в пределах зон производства продукции с высоким риском перекрестного загрязнения;
e) использование высокоэффективных методов очистки и обработки с целью исключения недостаточной очистки, часто являющейся причиной перекрестного загрязнения;
f) использование "закрытых систем" производства;
g) контроль наличия остатков предыдущего продукта или моющих средств и маркировка оборудования с указанием статуса чистоты.
5.20 Следует периодически проверять эффективность мер по предотвращению перекрестного загрязнения в соответствии с утвержденными инструкциями.
Аттестация (валидация)
5.21 Аттестация (валидация) направлена на повышение эффективности работы и проводится в соответствии с утвержденными методиками. Ее результаты должны оформляться документально.
5.22 При утверждении нового промышленного регламента или метода производства следует проверять их пригодность для серийного производства. Должно быть подтверждено, что данный процесс, используемые материалы и оборудование позволяют постоянно производить продукцию требуемого качества.
5.23 При существенных изменениях технологии, в т.ч. любых изменениях оборудования или материалов, способных влиять на качество продукции или воспроизводимость процесса, следует проводить аттестацию (валидацию) соответствующих процессов.
5.24 Для постоянного подтверждения достижения требуемых результатов следует проводить повторную аттестацию (валидацию) технологических процессов и методик.
Исходные материалы
5.25 Обеспечение исходными материалами является ответственной операцией, требующей полных сведений о поставщиках.
5.26 Исходные материалы следует закупать только у утвержденных поставщиков (указанных в спецификации) и, по возможности, непосредственно у производителей этих материалов. Требования к ним должны быть указаны в спецификациях на исходные материалы, утвержденных производителем лекарственных средств, и согласованы с поставщиком. Все вопросы, связанные с производством и контролем исходных материалов, в т.ч. работу с ними, маркировку, упаковку, порядок предъявления рекламаций и отклонения, следует согласовывать с производителем и поставщиком.
5.27 В каждой поставке следует проверять целостность тары, упаковки и пломб, соответствие накладной маркировке на таре (упаковке).
5.28 При поставке исходных материалов из нескольких серий каждую серию следует рассматривать как независимую в отношении отбора проб, проведения испытаний и получения разрешения на использование.
5.29 Исходные материалы, находящиеся на складе, должны быть маркированы соответствующим образом (5.13). Маркировка должна содержать, как минимум, следующую информацию:
- обозначение исходного продукта и внутризаводской код;
- номер серии, присвоенный при приемке;
- статус материала (например, "Карантин", "Испытания", "Разрешено", "Отклонено" и т.п.);
- срок годности или дату, после которой необходим повторный контроль.
Если складское хозяйство полностью оснащено компьютерами, указывать всю информацию на маркировке необязательно.
5.30 Контроль подлинности содержимого каждой упаковки с исходными материалами регламентируется соответствующими инструкциями и методиками. Упаковки с нерасфасованной продукцией, из которых были отобраны пробы, должны иметь соответствующую маркировку (6.13).
5.31 При производстве лекарственных средств могут использоваться только исходные материалы с неистекшим сроком годности и допущенные отделом контроля качества.
5.32 Исходные материалы должны выдаваться только специально назначенными лицами в соответствии с письменной инструкцией, при этом должны выполняться требования по точности взвешивания и отмеривания материалов в чистую и маркированную тару.
5.33 Следует выполнять независимую проверку каждого выданного вещества, его массы и объема. Результаты проверки должны быть оформлены документально.
5.34 Материалы, выданные для каждой серии, следует хранить в одном месте с четкой маркировкой.
Промежуточная и нерасфасованная продукция
5.35 Перед началом любой технологической операции следует принять меры, гарантирующие чистоту производственной зоны и оборудования, отсутствие любых исходных материалов, продукции, остатков продукции или документации, не относящихся к данному процессу.
5.36 Промежуточную и нерасфасованную продукцию следует хранить в надлежащих условиях.
5.37 Критические процессы должны быть аттестованы (валидированы) (5.21-5.24).
5.38 Выполнение всех необходимых операций по внутрипроизводственному контролю и контролю окружающей среды должно быть оформлено документально.
5.39 Все факты существенного отклонения выхода продукции от ожидаемого следует регистрировать и расследовать.
Упаковочные материалы
5.40 Закупке, хранению и контролю первичных и печатных упаковочных материалов следует уделять такое же внимание, как и исходному сырью.
5.41 Особое внимание следует уделять печатным материалам. Их следует хранить в безопасных условиях, исключающих доступ посторонних лиц. Разрезанные этикетки и другие разрозненные материалы должны храниться и транспортироваться раздельно в закрытой таре, исключающей их перепутывание. Разрешение на использование упаковочных материалов должно выдаваться только специально назначенными лицами в соответствии с утвержденной инструкцией.
5.42 Каждой поставке или серии первичных или печатных упаковочных материалов должен быть присвоен номер или отличительный знак.
5.43 Просроченные или непригодные к использованию печатные или первичные упаковочные материалы должны быть уничтожены с оформлением протокола.
Операции по упаковке
5.44 При упаковке продукции должен быть сведен к минимуму риск перекрестного загрязнения, перепутывания или подмены. Не допускается упаковывать продукцию различных видов в непосредственной близости друг от друга при отсутствии физического разделения зон.
5.45 Перед началом операций по упаковке следует убедиться в том, что рабочая зона, упаковочные линии, печатные машины и другое оборудование находятся в чистом состоянии и не содержат материалов, продукции или документов, относящихся к предшествующей работе и не использующихся в текущем процессе. Очистка линии упаковки продукции должна выполняться по специальным инструкциям.
5.46 Наименование и номер серии упаковываемой продукции должны быть указаны на каждой линии или установке.
5.47 При поступлении продукции и упаковочных материалов на участок упаковки следует проверить их количество, подлинность и соответствие инструкциям по упаковке.
5.48 Первичная упаковка для наполнения перед началом операции наполнения должна быть чистой. Особое внимание следует обратить на удаление любых загрязняющих веществ (осколков стекла, металлических частиц и пр.).
5.49 После наполнения и укупоривания продукции ее маркировку следует выполнять как можно быстрее. Если это невозможно, то следует принять необходимые меры против перепутывания продукции или ошибочной маркировки.
5.50 Правильность выполнения любых печатных операций (например, нанесения кодов или срока годности) при упаковке и после нее тщательно контролируется и оформляется документально. Следует уделять внимание ручной маркировке, которая должна контролироваться через регулярные интервалы времени.
5.51 Особые меры предосторожности должны приниматься при использовании разрезанных этикеток и нанесении печати вне линии упаковки. Для предотвращения перепутывания печатного материала рекомендуется использовать этикетки в рулоне вместо разрезанных этикеток.
5.52 Следует контролировать правильность работы электронных считывателей кодов, счетчиков этикеток и других подобных устройств.
5.53 Маркировка упаковочных материалов, нанесенная печатью или тиснением, должна быть отчетливой, устойчивой к воздействию света (выгоранию) и удалению.
5.54 При контроле процесса упаковки продукции на линии следует проверять, как минимум, следующее:
a) общий вид упаковки;
b) комплектность упаковки;
c) правильность использования упаковочных материалов в соответствии с документацией на данную продукцию;
d) правильность нанесения печатных надписей;
e) правильность работы устройств контроля на линии.
Образцы продукции, взятые с упаковочной линии, повторно на линию не возвращаются.
5.55 Если при упаковке продукции возникли непредвиденные обстоятельства, она может быть возвращена в производство только после специальной проверки, проведения расследования и с разрешения лица, имеющего на это полномочия. Указанные действия должны быть оформлены протоколами, которые следует хранить в установленном порядке.
5.56 При существенном или необычном расхождении, установленном при сопоставлении количества нерасфасованной продукции, печатных упаковочных материалов и количества единиц полученной готовой продукции, следует провести расследование и установить причину этого расхождения до получения разрешения на реализацию данной продукции.
5.57 После завершения операций по упаковке любые оставшиеся упаковочные материалы с нанесенным номером серии должны быть уничтожены с составлением протокола. Возврат на склад упаковочных материалов с отсутствующим номером серии выполняется в соответствии с утвержденной инструкцией.
Готовая продукция
5.58 До выдачи разрешения на реализацию готовая продукция должна содержаться под карантином в условиях хранения, установленных производителем.
5.59 Порядок оценки качества готовой продукции и требования к документации, необходимые для получения разрешения на реализацию, приведены в разделе 6.
5.60 После выдачи разрешения на реализацию готовая продукция хранится на складе готовой продукции в условиях, установленных производителем.
Отклоненные, повторно использованные и возвращенные материалы
5.61 Отклоненные материалы и продукция должны иметь четкую маркировку и храниться раздельно в зонах с ограниченным доступом. Они подлежат возврату поставщику, переработке (если это допустимо) или уничтожению. Любые выполненные действия должны документально оформляться и утверждаться специально назначенным персоналом.
5.62 Переработка отклоненной продукции допускается в исключительных случаях при условии отсутствия снижения качества готовой продукции и выполнения всех требований спецификаций. Переработка осуществляется в соответствии с утвержденными инструкциями после оценки возможного риска с последующим документальным оформлением.
5.63 Повторное использование всей (или части) ранее произведенной серии продукции требуемого качества путем объединения ее с другой серией того же продукта на определенном этапе производства допускается только после получения предварительного разрешения, подписанного ответственными лицами. Повторное использование продукции допускается только после оценки возможного риска (в т.ч. его влияния на срок годности серии) по утвержденной инструкции с оформлением протокола.
5.64 Необходимость дополнительного контроля готовой продукции, прошедшей переработку, или продукции, в которую был включен ранее изготовленный продукт, определяется отделом контроля качества.
5.65 Возвращенная с рынка продукция, над которой был утрачен контроль со стороны производителя, должна быть уничтожена, если не подтверждено соответствие ее качества заданным требованиям. Решение о повторной продаже, перемаркировке или регенерации может быть принято только после специального анализа, проведенного отделом контроля качества в соответствии с письменной инструкцией. При этом необходимо учитывать характер продукции, ее предысторию и состояние, соблюдение специальных условий хранения и время, прошедшее с даты выпуска. При любых сомнениях в отношении качества продукции не допускается ее повторное использование или повторный выпуск, но допускается ее химическая переработка с целью регенерации активных ингредиентов. Все выполняемые действия должны тщательно протоколироваться.
6 Контроль качества
Принципы
Контроль качества связан с отбором проб, проведением испытаний и проверок на соответствие требованиям спецификаций, инструкций и других документов, с организацией работы, документированием и процедурами выдачи разрешений на реализацию. Цель контроля качества - не допустить к использованию или реализации материалы или продукцию, не удовлетворяющие требованиям качества. Служба контроля качества выполняет исследования, проверки и участвует в принятии любых решений, касающихся качества продукции. Основополагающим принципом обеспечения контроля качества является независимость отдела контроля качества (раздел 1).
Общие положения
6.1 На каждом предприятии, выпускающем лекарственные средства, должен быть отдел контроля качества, независимый от других подразделений. Руководитель этого отдела должен иметь необходимый опыт и квалификацию. К отделу контроля качества относятся одна или несколько контрольных лабораторий. Для выполнения своих функций отдел должен быть обеспечен всеми необходимыми ресурсами.
6.2 Основные обязанности начальника отдела контроля качества изложены в разделе 2. На отдел возлагаются также обязанности по разработке, аттестации (валидации), внедрению всех инструкций (методик) по контролю качества; хранению контрольных образцов материалов и продукции; контролю правильности маркировки упаковок с материалами и продукцией; обеспечению контроля стабильности продукции; участию в расследовании рекламаций, связанных с качеством продукции, и т.п. Все эти функции должны выполняться в соответствии с утвержденными инструкциями с оформлением протоколов (при необходимости).
6.3 При оценке качества готовой продукции следует рассматривать все сопутствующие факторы, в т.ч. условия производства, результаты внутрипроизводственного контроля, анализ производственной документации (в т.ч. документации на упаковку), соответствие спецификациям на готовую продукцию и состояние окончательной упаковки готовой продукции.
6.4 Сотрудники отдела контроля качества должны иметь доступ в производственные зоны для отбора проб и проведения анализа.
Организация работы контрольных лабораторий
6.5 Помещения и оборудование контрольных лабораторий должны соответствовать общим и специальным требованиям, предъявляемым к зонам контроля качества (раздел 3).
6.6 Персонал, помещения и оборудование лабораторий должны соответствовать виду и объему производства. В отдельных случаях допускается использование сторонних лабораторий при условии выполнения ими требований, изложенных в разделе 7, и внесения соответствующих записей в протоколы контроля качества.
Документация
6.7 Документация контрольных лабораторий должна соответствовать требованиям, изложенным в разделе 4. К документации по контролю качества относятся:
- спецификации;
- методики отбора проб;
- методики и протоколы проведения испытаний (в т.ч. аналитические операционные листы и/или лабораторные журналы);
- аналитические отчеты и/или паспорта;
- результаты контроля окружающей среды в производственных помещениях;
- протоколы аттестации (валидации) аналитических методов;
- методики и протоколы калибровки (поверки) приборов и обслуживания аппаратуры.
Эта информация всегда должна быть готова к представлению в отдел контроля качества.
6.8 Документация по контролю качества, относящаяся к протоколам серий продукции, должна храниться в течение одного года после истечения срока годности серии.
6.9 Для некоторых данных (например, результатов аналитических испытаний, выходов готовой продукции, параметров окружающей среды и т.п.) целесообразно хранить протоколы в виде, позволяющем оценивать тенденции изменения параметров.
6.10 В дополнение к протоколам серий продукции следует хранить в доступном виде и другую первичную информацию (например, лабораторные журналы и/или протоколы).
Отбор проб
6.11 Отбор проб должен проводиться в соответствии с утвержденными инструкциями, включающими в себя:
- методику отбора проб;
- перечень используемого оборудования;
- количество отбираемых проб;
- инструкции по разделению отобранной пробы на части (при необходимости);
- тип и характеристики тары для отбора проб;
- нанесение маркировки на тару с отобранными пробами;
- специальные меры предосторожности, особенно относительно стерильных и опасных веществ;
- условия хранения;
- инструкции по очистке и хранению оборудования для отбора проб.
6.12 Отобранные контрольные образцы должны представлять репрезентативную выборку серии материалов или продукции. Возможен отбор проб, характеризующих критические стадии технологического процесса (например, его начало или окончание).
6.13 На маркировке тары с отобранными пробами должны быть указаны ее содержимое, даты отбора проб и упаковки, из которых эти пробы были отобраны.
6.14 Отобранные пробы каждой серии готовой продукции должны храниться в течение одного года после истечения срока ее годности. Готовая продукция должна, как правило, храниться в своей окончательной упаковке и в рекомендованных условиях. Контрольные образцы исходного сырья (кроме растворителей, газов и воды) должны храниться не менее двух лет после выдачи разрешения на реализацию продукции, если это допускается их стабильностью. При меньшем периоде стабильности, указанном в соответствующей спецификации, время хранения может быть сокращено. Количество контрольных образцов исходного сырья, материалов и продукции должно быть достаточным для проведения их полного повторного контроля.
Примечание - При необходимости срок хранения образцов исходного сырья должен быть не менее срока хранения соответствующей готовой продукции.
Проведение испытаний
6.15 Аналитические методики должны быть аттестованы (валидированы). Все испытания, приведенные в нормативной документации, должны быть выполнены в соответствии с утвержденными методиками.
6.16 Полученные результаты испытаний должны оформляться документально с тщательной проверкой всех внесенных данных. Все расчеты должны тщательно проверяться.
6.17 Проводимые испытания следует оформлять документально с указанием:
a) наименования материала или продукции и дозированной формы;
b) номера серии и наименования производителя и/или поставщика;
c) ссылки на соответствующие спецификации и методики испытаний;
d) результатов испытаний, в т.ч. наблюдения, вычисления и ссылки на все паспорта анализов;
e) даты проведения испытаний;
f) фамилий и инициалов лиц, проводивших испытание;
g) фамилий и инициалов лиц, подтверждавших проведение испытаний и результаты вычислений;
h) однозначного заключения о выдаче разрешения или отклонении продукции (или другого решения о статусе продукции), даты и подписи ответственного лица.
6.18 Все операции по внутрипроизводственному контролю, в т.ч. операции, выполняемые лицами, непосредственно работающими в производственных зонах, должны проводиться в соответствии с методиками, утвержденными отделом контроля качества, а их результаты оформляться документально.
6.19 Особое внимание следует уделять качеству лабораторных реактивов, мерной лабораторной посуды и титрованных растворов, стандартных образцов и питательных сред. Они должны готовиться в соответствии с письменными инструкциями.
6.20 Реактивы, предназначенные для длительного использования, должны иметь маркировку с указанием даты приготовления и с подписями исполнителей. На этикетке должен быть указан срок годности нестабильных реактивов, питательных сред и специфические условия их хранения. Для титрованных растворов следует указывать дату последнего установления титра и соответствующий поправочный коэффициент.
6.21 При необходимости на таре следует указывать дату получения каждого вещества, используемого для проведения испытаний (например, реактивов и стандартных образцов) с соответствующими инструкциями по их использованию и хранению. В некоторых случаях после получения или перед использованием реактива может возникнуть необходимость проведения его испытания на подлинность и/или другого испытания.
6.22 Животных, используемых для контроля компонентов, материалов или продукции, следует, при необходимости, помещать в карантин перед работой с ними. Уход и контроль за животными должны быть организованы так, чтобы обеспечить их пригодность к использованию по назначению. Животные должны быть маркированы, истории работы с ними должны быть оформлены документально.
7 Работа по контрактам на производство продукции и проведение анализов
Принципы
Во избежание разночтений, способных привести к ухудшению качества продукции или выполнения работ, следует тщательно составлять, согласовывать и контролировать выполнение контрактов на производство продукции и проведение анализов. Контракт между заказчиком и исполнителем должен быть составлен в письменной форме с указанием четко определенных обязанностей каждой из сторон. Контракт должен устанавливать порядок действий и ответственность Уполномоченного лица за выдачу разрешения на реализацию каждой серии продукции.
Примечание - Настоящий раздел рассматривает только ответственность производителей за выполнение требований настоящего стандарта.
Общие положения
7.1 Контракт должен быть составлен в письменной форме и должен включать в себя перечень производственных операций и/или анализов, выполняемых на основании контракта, и проводимых технических мероприятий.
7.2 Выполнение контракта на производство и/или проведение анализов, в т.ч. с учетом предложенных изменений технического или другого характера, должно соответствовать требованиям нормативных документов на производство и регистрационного досье на данную продукцию.
Заказчик
7.3 Заказчик несет ответственность за оценку компетентности исполнителя и его способность выполнить контракт надлежащим образом, а также за включение в контракт положений, обеспечивающих выполнение требований настоящего стандарта.
7.4 Заказчик предоставляет исполнителю информацию, необходимую для выполнения предусмотренных в контракте работ в соответствии с требованиями регистрационного досье, нормативных документов и законодательства. Заказчик должен убедиться в том, что исполнитель обладает полной информацией о факторах, связанных с продукцией или работой, выполняемой по контракту, и представляющих опасность для его помещений, оборудования, персонала, других материалов или продуктов.
7.5 Заказчик должен убедиться в том, что вся готовая продукция и материалы, поставленные ему исполнителем, соответствуют спецификациям, а разрешение на реализацию этой продукции выдано Уполномоченным лицом.
Исполнитель
7.6 Исполнитель должен иметь соответствующие помещения, оборудование, знания, опыт и компетентный персонал для надлежащего выполнения контракта. Контракт на производство может выполнять исполнитель, имеющий разрешение на производство лекарственных средств.
7.7 Исполнитель должен гарантировать пригодность получаемых им материалов и продукции для использования по назначению.
7.8 Исполнитель не должен привлекать третью сторону для выполнения работ или их части, порученных ему по контракту, без предварительного рассмотрения и согласования с заказчиком. При заключении соглашения между исполнителем и третьей стороной должна быть гарантия того, что информация о производстве и проведении анализов будет доступна заказчику в соответствии с контрактом.
7.9 Исполнитель не должен предпринимать действия, которые могут оказать отрицательное влияние на качество продукции, произведенной и/или проанализированной им в соответствии с контрактом.
Контракт
7.10 Контракт заключается между заказчиком и исполнителем. В контракте указывается ответственность сторон за производство и контроль продукции. Технические аспекты контракта должны быть составлены компетентными лицами, обладающими соответствующими знаниями в области технологии производства лекарственных средств, проведения анализов и требований настоящего стандарта. Все соглашения по производству и проведению анализов должны быть согласованы обеими сторонами и должны соответствовать требованиям регистрационного досье.
7.11 В контракте должно быть указано, каким образом Уполномоченное лицо, ответственное за выпуск серии продукции для реализации, гарантирует, что каждая серия продукции была изготовлена и проконтролирована в соответствии с требованиями нормативной документации и регистрационного досье.
7.12 В контракте должны быть указаны лица, ответственные за закупку, проведение испытаний и выдачу разрешения на использование материалов, производство, контроль качества (в т.ч. внутрипроизводственный контроль), отбор проб и проведение анализов. При заключении контракта на анализ продукции должна быть предусмотрена возможность проведения отбора проб исполнителем в помещениях производителя продукции.
7.13 Протоколы производства, анализов и реализации продукции, соответствующие контрольные образцы должны храниться у заказчика или должны быть ему доступны. Перечень документов, относящихся к оценке качества продукции и связанных с рекламациями или возникновением сомнений в качестве продукции, должен быть определен заказчиком в инструкциях по отклонению (отзыву) продукции. Эти документы должны быть доступны для заказчика.
7.14 В контракте должно быть предусмотрено право заказчика на посещение производства исполнителя.
7.15 При заключении контракта на проведение анализов возможно инспектирование исполнителя надзорными органами.
8 Рекламации и отзыв продукции
Принципы
Все рекламации и информация, касающиеся продукции с предполагаемыми нарушениями качества, должны быть тщательно проанализированы в соответствии с инструкциями. На предприятии должна быть создана система быстрого и эффективного отзыва с рынка продукции с явными или предполагаемыми нарушениями качества в следующих случаях:
a) продукция оказалась опасной при обычных условиях применения;
b) продукция терапевтически неэффективна;
c) по качественному и количественному составу продукция не соответствует составу, указанному в регистрационном досье;
d) контроль готового лекарственного средства и/или ингредиентов, контроль промежуточных стадий производственного процесса не проводился или не выполнялись требования или обязательства, относящиеся к условиям выдачи разрешения на производство лекарственных средств;
e) при других непредвиденных обстоятельствах.
Рекламации
8.1 На предприятии должен быть назначен сотрудник, имеющий в распоряжении необходимый персонал, ответственный за рассмотрение рекламаций и принятие решений. Если этот сотрудник не является Уполномоченным лицом, то Уполномоченное лицо должно быть поставлено в известность о всех фактах рекламаций, расследований и отзывов продукции.
8.2 Действия по рассмотрению рекламаций на предположительно некачественную продукцию и принятие решения об отзыве продукции должны быть изложены в соответствующей инструкции.
8.3 Следует обеспечить тщательность расследования рекламации и его документальное оформление с описанием деталей любой претензии на качество продукции. К этой работе обычно привлекается лицо, ответственное за контроль качества продукции.
8.4 Если обнаружено или подозревается несоответствие качества какой-либо серии продукции, следует принять решение о проверке аналогичных серий. В частности, проверке могут подлежать другие серии, содержащие переработанный продукт из этой серии.
8.5 Решения и меры, принятые по любой рекламации, должны быть внесены в соответствующий протокол на серию продукции.
8.6 Протоколы рекламаций следует регулярно рассматривать и анализировать с целью выявления специфических и повторяющихся факторов, требующих особого внимания и способствующих отзыву продукции.
8.7 При ухудшении качества продукции или других проблемах с качеством, в отношении которых предпринимаются корректирующие действия, производитель обязан известить соответствующие компетентные органы.
Отзыв продукции
8.8 На предприятии должен быть назначен сотрудник, имеющий в распоряжении необходимый персонал, ответственный за своевременный отзыв продукции с рынка. Как правило, это лицо должно быть независимым от служб реализации и маркетинга. Если этот сотрудник не является Уполномоченным лицом, то Уполномоченное лицо должно быть осведомлено обо всех фактах отзывов продукции.
8.9 Порядок отзыва продукции должен быть регламентирован инструкцией, которую следует регулярно проверять и пересматривать.
8.10 Отзыв продукции должен осуществляться оперативно и в любое время.
8.11 Компетентные органы стран, куда могла быть направлена продукция, должны быть немедленно информированы о принятии решения об отзыве продукции в связи с подозрением или обнаружением несоответствия ее качества.
8.12 Документация по отгрузке продукции должна быть доступной для лица (лиц), ответственного(ых) за отзыв продукции, и содержать достаточную информацию об оптовых покупателях и прямых заказчиках (адреса, номера телефонов/факсов в рабочее и в нерабочее время, номера серий и объемы поставок), экспортных поставках и поставках образцов лекарственных средств.
8.13 На отозванной продукции должна быть соответствующая маркировка. Отозванную продукцию следует хранить в надежно изолированных зонах до принятия решения о ее дальнейшем использовании или утилизации.
8.14 Последовательность действий при отзыве продукции должна быть оформлена документально. Окончательный отчет должен содержать баланс между количеством поставленной и отозванной продукции.
8.15 Эффективность мероприятий по отзыву продукции следует регулярно анализировать.
9 Самоинспекция
Принципы
Самоинспекция должна проводиться с целью проверки выполнения предприятием требований настоящего стандарта и принятия необходимых мер по устранению недостатков.
9.1 С целью выполнения принципов обеспечения качества вопросы, связанные с работой персонала, содержанием помещений, эксплуатацией оборудования, документацией, производством, контролем качества и реализацией лекарственных средств, мероприятиями по работе с рекламациями и отзыву продукции, а также проведению самоинспекций, должны регулярно рассматриваться в соответствии с утвержденной программой.
9.2 Самоинспекция должна проводиться независимо и тщательно специально назначенным лицом (лицами) из штата предприятия. Полезно проводить независимый аудит экспертами сторонних организаций.
9.3 Результаты проведения самоинспекций должны быть оформлены документально. Протоколы, составленные по результатам проведения самоинспекции, должны включать в себя всю полученную информацию и необходимые корректирующие действия (при необходимости). Действия, принимаемые по результатам самоинспекции, следует оформлять документально.
Приложение 1
Производство стерильных лекарственных средств
Принципы
К производству стерильных лекарственных средств предъявляются особые требования, направленные на сведение к минимуму риска загрязнения микроорганизмами, частицами и пирогенами. Выполнение этих требований во многом зависит от опыта персонала, его подготовки и отношения к работе. Особенно высокие требования предъявляются к обеспечению качества, подготовке и выполнению технологических процессов, их тщательной отработке и аттестации (валидации). Контроль конечной стадии производства или контроль готового продукта не может рассматриваться как единственное средство обеспечения стерильности или других показателей качества продукции.
Примечание - Настоящий стандарт не устанавливает методы определения чистоты воздуха, поверхностей и пр. по микроорганизмам и частицам. Эти требования приводятся в других нормативных документах (стандартах СЕN, ИСО, ГОСТ Р ИСО и др.).
Общие положения
1 Производство стерильных препаратов должно быть организовано в чистых помещениях (зонах) с воздушными шлюзами для обеспечения доступа персонала и/или перемещения оборудования и материалов. В чистых помещениях (зонах) должен поддерживаться уровень чистоты по соответствующему стандарту, а воздух должен подаваться через фильтры необходимой эффективности.
2 Подготовка первичной упаковки, приготовление продукта и наполнение должны выполняться в отдельных чистых зонах (помещениях).
Процессы производства стерильных лекарственных средств подразделяются на две категории:
- предусматривающие финишную стерилизацию (т.е. стерилизацию в герметичной первичной упаковке);
- проводимые в асептических условиях на одном или всех этапах производства.
3 Чистые помещения (зоны) для производства стерильной продукции классифицируются в соответствии с требованиями к окружающей среде с целью сведения к минимуму риска загрязнения продукта или материалов частицами или микроорганизмами. Каждая производственная операция требует определенного уровня чистоты окружающей среды в эксплуатируемом состоянии.
Для обеспечения соответствия чистых помещений (чистых зон) требованиям, предъявляемым к эксплуатируемому состоянию, их проект должен предусматривать достижение заданных классов чистоты воздуха в оснащенном состоянии.
Оснащенное состояние - состояние, в котором чистое помещение функционирует, технологическое оборудование полностью укомплектовано, но персонал отсутствует.
Эксплуатируемое состояние - состояние чистого помещения, в котором технологическое оборудование функционирует в требуемом режиме с заданным числом работающего персонала.
Чистые зоны при производстве стерильных лекарственных средств подразделяются на четыре типа:
А - локальная зона для проведения операций, представляющих высокий риск для качества продукции, например, зоны наполнения, укупорки, вскрытия ампул и флаконов, соединения частей оборудования в асептических условиях. Как правило, в таких зонах используется однонаправленный (ламинарный) поток воздуха, обеспечивающий в незамкнутой чистой зоне однородную скорость 0,36-0,54 м/с (рекомендуемое значение). Поддержание однонаправленности воздушного потока должно быть подтверждено при аттестации (валидации). В закрытых изолирующих устройствах и перчаточных боксах можно использовать однонаправленный поток воздуха с меньшей скоростью;
В - зона, непосредственно окружающая зону А и предназначенная для асептического приготовления и наполнения;
С и D - чистые зоны для выполнения менее ответственных стадий производства стерильной продукции.
Классификация зон по загрязнению воздуха частицами приведена в таблице.
|
Тип зоны |
Максимально допустимое число частиц в 1 м | |||
|
в оснащенном состоянии (b) |
в эксплуатируемом состоянии (b) | |||
|
0,5 мкм (d) |
5 мкм |
0,5 мкм (d) |
5 мкм | |
|
А |
3500 |
1 (е) |
3500 |
1 (е) |
|
В (с) |
3500 |
1 (е) |
350000 |
2000 |
|
С (с) |
350000 |
2000 |
3500000 |
20000 |
|
D (c) |
3500000 |
20000 |
Не регламентируется (f) |
Не регламентируется (f) |
|
Примечания | ||||
|
(a) Концентрация частиц с размерами, равными или превышающими указанные значения, определяется с помощью дискретного счетчика аэрозольных частиц. | ||||
|
В зоне А следует предусматривать непрерывный контроль концентрации частиц. Рекомендуется предусматривать такой контроль и в зоне В*. | ||||
|
При текущем контроле зон А и В отбираются пробы общим объемом не менее 1 м | ||||
|
(b) Уровень загрязнения частицами, показанный в таблице для оснащенного состояния, должен достигаться через 15-20 мин (рекомендуемое значение времени восстановления) после завершения процесса при отсутствии персонала. Уровень загрязнения частицами для зоны А в эксплуатируемом состоянии должен поддерживаться в зоне, непосредственно окружающей продукт, когда продукт или открытая упаковка имеют прямой контакт с окружающей средой. | ||||
|
Допускается, что в процессе наполнения не всегда может быть показано соответствие стандартам по частицам, поскольку сам продукт может выделять частицы или капельки. | ||||
|
(c) В зонах В, С и D кратность воздухообмена должна определяться с учетом размеров помещения, находящегося в нем оборудования и персонала. Для зон А, В и С система подготовки воздуха должна иметь соответствующие фильтры (типа НЕРА). | ||||
|
(d) Значения максимально допустимого числа аэрозольных частиц с размерами 0,5 мкм и более в оснащенном и эксплуатируемом состояниях ориентировочно соответствуют классам чистоты по ГОСТ ИСО 14644-1. | ||||
|
(e) Предполагается, что в воздухе этих зон частицы с размерами 5 мкм и более должны отсутствовать. Поскольку невозможно статистически достоверно доказать полное отсутствие частиц, для этих случаев установлен предел - 1 частица/м | ||||
|
(f) Требования к этой зоне и допустимые пределы зависят от характера выполняемых в ней операций. | ||||
|
________________ | ||||
|
- непрерывный контроль концентрации частиц может быть заменен на периодический контроль, проводимый через небольшие интервалы времени, например, перед началом смены и после ее окончания; | ||||
|
- вместо отбора пробы с общим объемом не менее 1 м | ||||
 воздуха,
воздуха,
Требования к другим параметрам (температуре, относительной влажности и др.) зависят от продукта и характера технологических операций. Эти параметры не связаны с классами чистоты.
Примеры операций, выполняемых в зонах различных типов, приведены в таблицах (пункты 11 и 12):
|
Тип зоны |
Примеры операций для продуктов, подлежащих финишной стерилизации (пункт 11) |
|
А |
Наполнение продуктом, когда его нельзя подвергать риску загрязнения |
|
С |
Приготовление растворов, когда их нельзя подвергать риску загрязнения |
|
D |
Приготовление растворов и подготовка первичной упаковки, материалов и др. для последующего наполнения |
|
Тип зоны |
Примеры операций для асептического приготовления (пункт 12) |
|
А |
Асептическое приготовление и наполнение |
|
С |
Приготовление растворов, подлежащих фильтрации |
|
D |
Операции с материалами после мойки |
4 Следует предусматривать контроль чистоты зон различных типов по частицам в эксплуатируемом состоянии.
5 В асептическом производстве необходимо достаточно часто проводить микробиологический контроль с использованием методов седиментации на чашки, отбора проб в объеме воздуха и с поверхностей (например, смывы и контактные пластины). Методы отбора проб, используемые в эксплуатируемом состоянии, не должны вносить помех в защиту зоны. Результаты контроля следует учитывать при рассмотрении документации на серию готовой продукции. После выполнения критических операций следует проводить микробиологический контроль поверхностей и персонала.
Следует также проводить дополнительный микробиологический контроль вне технологического процесса, например, после аттестации (валидации) оборудования, выполнения очистки и дезинфекции.
Рекомендуемые пределы допустимого микробного загрязнения чистых зон в эксплуатируемом состоянии приведены в таблице.
|
Тип зоны |
Рекомендуемые пределы микробного загрязнения (а) | |||
|
в воздухе, КОЕ/м |
седиментация |
контактные пластины диаметром 55 мм, КОЕ/пластина |
отпечаток перчатки | |
|
А |
< 1 |
<1 |
<1 |
<1 |
|
В |
10 |
5 |
5 |
5 |
|
С |
100 |
50 |
25 |
- |
|
D |
200 |
100 |
50 |
- |
|
Примечания | ||||
|
(а) Указаны средние значения. | ||||
|
(b) Допускается экспонирование отдельных седиментационных пластин менее 4 ч. | ||||
6 В зависимости от результатов проводимого контроля следует установить пределы предупреждения и действия для показателей загрязнения частицами и микроорганизмами, а также предусмотреть выполнение корректирующих действий в случае превышения этих пределов.
Изолирующая технология
7 Применение изолирующей технологии сводит к минимуму влияние человека на технологические зоны. В асептическом производстве это позволяет значительно снизить риск микробного загрязнения продукции из окружающей среды.
В изоляторах и передаточных устройствах всех типов должны выполняться установленные требования к качеству воздуха. При этом следует учитывать, в какой степени возможны утечки (повреждения), вызванные особенностями конструкции или материалов изолятора. Передаточные устройства могут быть разными: от конструкций с одинарной или двойной дверью до полностью герметизированных систем, предусматривающих проведение стерилизации.
Процесс передачи материалов внутрь и наружу изолятора является одним из наиболее сильных потенциальных источников загрязнений. Пространство внутри изолятора предназначено для проведения операций, представляющих высокий риск для качества продукции. В то же время допускается организация рабочих зон внутри изолятора без однонаправленного (ламинарного) потока воздуха. Требования к чистоте воздуха в среде, окружающей изолятор, зависят от конструкции изолятора и его назначения. Эта среда должна контролироваться, и для асептического производства соответствовать, по крайней мере, зоне D.
8 Изоляторы могут быть введены в эксплуатацию только после завершения аттестации (валидации), которая должна учитывать все критические факторы изолирующей технологии, например, качество воздуха внутри и снаружи изолятора, порядок обработки изолятора, технологию передачи и целостность изолятора.
9 Следует установить порядок текущего контроля, включающий в себя достаточно частое проведение испытаний на наличие утечек в изоляторе и системе "перчатки-рукава".
Технология "выдувание-наполнение-герметизация"
10 Устройство "выдувание-наполнение-герметизация" - оборудование специальной конструкции, в котором в течение одного непрерывного технологического цикла из термопластичного гранулята формируются упаковки, которые наполняются продуктом и герметизируются. Все эти операции проводятся в пределах одного автоматического комплекса. Оборудование "выдувание-наполнение-герметизация", используемое в асептическом производстве и имеющее зону А с эффективным потоком воздуха, может быть установлено, по крайней мере, в зоне С при условии использования персоналом одежды, применяемой в зонах А и В. Окружающая среда в оснащенном состоянии должна соответствовать установленным требованиям по частицам и по микроорганизмам, а в эксплуатируемом состоянии - только по микроорганизмам. Оборудование "выдувание-наполнение-герметизация", используемое в производстве продуктов, подлежащих финишной стерилизации, должно устанавливаться, по крайней мере, в зоне D.
Особое внимание следует обратить на следующее:
- конструкцию и аттестацию оборудования;
- аттестацию и воспроизводимость процессов "очистка на месте" и "стерилизация на месте";
- параметры чистого помещения, в котором установлено оборудование;
- обучение операторов и их одежду;
- действия в критической зоне оборудования, в т.ч. выполнение подсоединений и сборки в асептических условиях до начала наполнения.
Продукты, подлежащие финишной стерилизации
11 Подготовка первичной упаковки, материалов и большинства продуктов должна проводиться, по крайней мере, в зоне D, чтобы обеспечить достаточно низкий уровень загрязнения микроорганизмами и частицами перед стадиями фильтрации и стерилизации. При повышенном риске загрязнения микроорганизмами (например, когда продукт, являющийся хорошей питательной средой для микроорганизмов, должен храниться в течение длительного времени до стерилизации или он обычно готовится в открытых емкостях) технологические операции следует проводить в окружающей среде типа С.
Наполнение продуктами, подлежащими финишной стерилизации, должно проводиться, по крайней мере, в зоне С.
При повышенном риске загрязнения продукта от окружающей среды, например, если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации, наполнение должно проводиться в зоне А, находящейся в окружающей среде типа С. Приготовление и наполнение мазями, кремами, суспензиями и эмульсиями перед финишной стерилизацией следует, как правило, проводить в окружающей среде типа С.
Асептическое производство
12 Операции с материалами после мойки должны проводиться, по крайней мере, в окружающей среде типа D. Операции со стерильными исходными материалами и компонентами, если на последующих стадиях процесса не предусмотрена их стерилизация или фильтрация через фильтры, удерживающие микроорганизмы, должны проводиться в зоне А, находящейся в окружающей среде типа В.
Приготовление растворов, которые в ходе технологического процесса подлежат стерилизующей фильтрации, должно проводиться в зоне С. Если фильтрация не предусмотрена, приготовление материалов и продуктов должно проводиться в зоне А, которая находится в окружающей среде типа В.
Операции по переработке и наполнению приготовленных в асептических условиях продуктов следует проводить в зоне А, находящейся в окружающей среде типа В.
Транспортирование частично закрытых первичных упаковок, например, при лиофильной сушке, должно до завершения укупорки проводиться либо в зоне А, находящейся в окружающей среде типа В, либо в герметичных передаточных устройствах в окружающей среде типа В.
Приготовление и наполнение стерильных мазей, кремов, суспензий и эмульсий должно проводиться в зоне А, находящейся в окружающей среде типа В, в том случае, когда продукт не защищен от окружающей среды и не подлежит последующей фильтрации.
Персонал
13 В чистых зонах должно находиться минимально необходимое количество персонала. Это особенно важно для асептического производства. Проверки и контрольные операции следует, по возможности, проводить, находясь за пределами чистых зон.
14 Весь персонал (в т.ч. персонал, занятый очисткой и техническим обслуживанием), работающий в таких зонах, должен проходить систематическое обучение по вопросам производства стерильных продуктов, включая гигиену и основы микробиологии. Следует обратить особое внимание на инструктаж и контроль за работниками, не прошедшими такого обучения, но которым необходимо входить в чистую зону (например, лицам, занятым в строительстве или техническом обслуживании).
15 Не допускается вход в зоны стерильного производства персонала, работающего с материалами из тканей животных или культурами микроорганизмов, которые не используются в текущем технологическом процессе, за исключением особых случаев, при которых необходимо соблюдение специальных инструкций для входа в эти зоны.
16 Необходимо выполнять требования к личной гигиене и чистоте. Персонал, занятый в производстве стерильных препаратов, должен знать порядок оповещения руководства (службы качества) о любых факторах, которые могут привести к повышению уровня загрязнения сверх допустимой нормы (как по количеству, так и по разновидностям). Следует организовать контроль за состоянием здоровья персонала. Решение о мерах в отношении персонала, который может стать источником микробного загрязнения, должно приниматься специально назначенным лицом.
17 Переодевание и мытье следует выполнять в соответствии с инструкциями, чтобы свести к минимуму риск загрязнения одежды, предназначенной для чистых зон, и внесения загрязнения в чистые зоны.
18 В чистых зонах персоналу запрещается носить наручные часы и ювелирные украшения, а также применять косметику.
19 Одежда и ее качество должны соответствовать технологическому процессу и типу зоны. Ее нужно носить так, чтобы обеспечить защиту продукта от загрязнений.
К одежде, предназначенной для зон различных типов, предъявляются следующие требования.
Зона D. Головной убор должен закрывать волосы. Борода также должна быть закрыта (специальной маской). Следует носить защитный костюм общего назначения, соответствующую обувь или бахилы, надеваемые поверх обуви. Должны быть приняты меры для предотвращения проникания любого загрязнения в чистую зону извне.
Зона С. Головной убор должен закрывать волосы. Борода и усы также должны быть закрыты. Следует носить костюм (цельный или состоящий из двух частей), плотно облегающий запястья, с воротником-стойкой и соответствующую обувь или бахилы. Одежда и обувь не должны выделять волокна или частицы.
Зоны А и В. Головной убор должен полностью закрывать волосы, а также бороду и усы (при их наличии). Края головного убора должны быть убраны под воротник костюма. Следует носить маску, чтобы предотвратить распространение капель, стерильные, неопудренные резиновые или полимерные перчатки и стерильные (или дезинфицированные) бахилы. Нижняя часть штанин должна быть заправлена внутрь бахил, а рукава одежды - в перчатки. Защитная одежда не должна выделять волокна или частицы и должна удерживать частицы, отделяющиеся от тела.
20 Наружная одежда не должна попадать в комнаты для переодевания, ведущие в зоны В и С. Каждый работник в зонах А и В должен быть обеспечен чистой стерильной одеждой на каждую смену. Во время работы перчатки следует регулярно дезинфицировать. Маски и перчатки следует менять, по крайней мере, каждую смену.
21 Одежда, предназначенная для чистой зоны, должна очищаться и храниться таким образом, чтобы исключить накопление загрязнений, которые могут от нее впоследствии отделиться. Эти операции следует выполнять в соответствии с инструкциями. Желательно иметь отдельные участки для подготовки такой одежды (прачечные). Одежда, подготовленная ненадлежащим образом, повреждает волокна ткани и увеличивает риск отделения частиц.
Помещения
22 Для того чтобы свести к минимуму отделение частиц или микроорганизмов, или их накопление, обеспечить возможность многократной обработки моющими и дезинфицирующими средствами, все открытые поверхности в чистых зонах должны быть гладкими, непроницаемыми, без трещин и изломов.
23 Чтобы уменьшить накопление пыли и облегчить очистку, в помещении не должно быть труднодоступных для очистки мест. Количество выступающих частей оборудования, полок и стеллажей должно быть минимальным. Конструкция дверей должна предусматривать отсутствие труднодоступных для очистки мест. По этой причине применение раздвижных дверей может быть нежелательным.
24 Подвесные потолки должны быть герметичными с целью предотвращения попадания загрязнения из пространства над ними.
25 Монтаж трубопроводов, воздуховодов и другого оборудования следует выполнять так, чтобы не было труднодоступных для очистки зон и поверхностей, а также негерметичных мест.
26 Запрещается устанавливать воронки и канализационные трубы в зонах А и В, используемых для асептического производства. В других зонах следует предусматривать разрыв струи между оборудованием и канализационной трубой (воронкой). При удалении стоков в чистых помещениях более низких классов следует предусматривать трапы (гидрозатворы) для предотвращения обратного потока.
27 Комнаты (помещения) для переодевания должны проектироваться по принципу воздушных шлюзов. Они должны обеспечивать физическое разделение различных этапов переодевания, чтобы сводить к минимуму загрязнение технологической одежды частицами и микроорганизмами, и эффективное обтекание помещений потоком отфильтрованного воздуха. Последняя часть комнаты (помещения) для переодевания в оснащенном состоянии должна иметь тот же класс чистоты, что и зона, в которую она ведет. В некоторых случаях целесообразно иметь отдельные комнаты (помещения) для переодевания для входа в чистые зоны и выхода из них. Устройства для мытья рук следует, как правило, устанавливать только в передней части комнаты (помещения) для переодевания.
28 Обе двери воздушного шлюза не должны быть одновременно открыты. Для предотвращения открывания более чем одной двери одновременно следует предусмотреть систему блокировки или оповещения (визуальную и/или звуковую).
29 Система обеспечения отфильтрованным воздухом должна поддерживать положительный перепад давления по отношению к окружающим зонам более низкого типа и соответствующий поток воздуха при всех условиях функционирования, а также эффективное обтекание воздухом контролируемой зоны. Соседние помещения различных типов должны иметь перепад давления 10-15 Па (рекомендуемое значение). Особое внимание следует уделять защите зон с большей степенью риска, т.е. непосредственному окружению открытого продукта или компонентов, контактирующих с продуктом. При работе с некоторыми материалами (патогенными, высокотоксичными, радиоактивными и пр.) или живыми вирусами, бактериями и препаратами из них могут потребоваться специальные способы подготовки воздуха и обеспечения перепада давления. Для некоторых операций может потребоваться деконтаминация оборудования и обработка удаляемого воздуха.
30 Следует наглядно показать, что направление воздушных потоков не представляет риска для загрязнения продукта, например, следует удостовериться, что в зону, представляющую наибольший риск для качества продукта, с воздушным потоком не поступают частицы, источниками выделения которых являются обслуживающий персонал, выполняемая операция или оборудование.
31 Следует предусмотреть систему аварийного оповещения об отказе системы подготовки воздуха, установить датчики перепада давления между зонами, там, где это имеет важное значение. Значения перепада давления необходимо регулярно записывать или документировать иным способом.
Оборудование
32 Ленты конвейеров не должны пересекать разделительный барьер между зонами А или В и рабочей зоной с меньшей чистотой воздуха, если только сама лента не подвергается непрерывной стерилизации (например, в туннеле стерилизации).
33 Конструкция, установка и расположение оборудования, фитингов (мест соединения) и зон обслуживания должны предусматривать возможность работы с оборудованием, его технического обслуживания и ремонта снаружи чистой зоны. В случае необходимости проведения стерилизации ее следует выполнять после максимально полной разборки оборудования (насколько это возможно).
34 Если при проведении технического обслуживания или ремонта оборудования, находящегося в чистой зоне, был нарушен уровень чистоты (стерильности), то перед возобновлением производства следует выполнять соответствующую очистку, дезинфекцию и/или стерилизацию этого оборудования (зоны).
35 Получение воды требуемого качества должно гарантироваться проектом, конструкцией, монтажом и техническим обслуживанием систем подготовки и распределения воды. Не допускается эксплуатация оборудования подготовки воды сверх проектной мощности. Приготовление, хранение и распределение воды для инъекций следует выполнять так, чтобы исключить рост микроорганизмов, например, за счет постоянной циркуляции воды при температуре выше плюс 70 °С.
36 Все критическое оборудование (стерилизаторы, системы подготовки и фильтрации воздуха, воздушные и газовые фильтры, системы приготовления, хранения и распределения воды и пр.) подлежат аттестации (валидации) и плановому техническому обслуживанию. Их повторный ввод в действие должен быть разрешен в установленном порядке.
Очистка и дезинфекция
37 Особое значение имеет обработка чистых помещений, которые должны быть тщательно очищены в соответствии с инструкцией. При проведении дезинфекции не следует ограничиваться применением только одного дезинфицирующего средства. Для обнаружения устойчивых штаммов микроорганизмов в помещении следует проводить регулярный контроль.
38 Следует контролировать микробное загрязнение моющих и дезинфицирующих средств. Растворы должны находиться в предварительно обработанных контейнерах. Хранение растворов, для которых не предусмотрена последующая стерилизация, допускается только в течение определенного периода времени. В зонах А и В следует применять только стерильные моющие и дезинфицирующие средства.
39 С целью уменьшения микробного загрязнения недоступных для очистки мест может использоваться газовая дезинфекция (фумигация).
Технологический процесс
40 На всех стадиях производства, в т.ч. на стадиях, предшествующих стерилизации, следует предусматривать меры по предупреждению загрязнения.
41 Не допускается приготовление лекарственных средств микробиологического происхождения или наполнение ими в зонах, используемых для производства других лекарственных средств. Операции наполнения вакцинами убитых организмов или экстрактами бактерий после их инактивации могут быть выполнены в тех же помещениях, что и операции наполнения другими стерильными лекарственными средствами.
42 При аттестации (валидации) асептического процесса следует проводить его имитацию с использованием питательной среды (наполнение средами). При выборе питательной среды следует учитывать дозированную форму продукта и селективность, прозрачность, концентрацию и пригодность этой питательной среды для стерилизации. Имитация процесса должна как можно ближе отражать текущий процесс асептического производства и включать в себя все критические стадии технологического процесса. Этот тест также должен учитывать различные воздействия на процесс, которые возникают в текущем производстве, а также ситуации "наихудшего случая".
Имитацию процесса с использованием питательных сред следует проводить при первоначальной аттестации процесса стерилизации, в ходе которой последовательно проводятся три успешных испытания. В дальнейшем следует повторять испытания через определенные интервалы времени, а также после любых существенных изменений в системе вентиляции и кондиционирования воздуха, оборудовании, процессе или количестве смен.
Как правило, испытания с использованием питательных сред следует повторять два раза в год для каждой смены и каждого процесса. Число первичных упаковок с питательной средой должно быть достаточным для получения достоверной оценки. Для малых серий число первичных упаковок с питательной средой должно быть равно размеру серии продукции. Испытания проводятся с целью проверки отсутствия роста микроорганизмов во всех упаковках, однако допустимым считается уровень загрязнения упаковок менее 0,1% с доверительным интервалом 0,95. Производитель должен установить уровни предупреждения и действия. Любой случай выявления загрязнения подлежит расследованию.
43 Проведение аттестации (валидации) не должно вредить технологическому процессу.
44 Источники водоснабжения, оборудование подготовки воды и приготовленная вода подлежат регулярному контролю на наличие химических и биологических загрязнений и, в необходимых случаях, на эндотоксины. Должна быть организована система документирования результатов контроля и любых предпринимаемых действий.
45 В чистых зонах, особенно в ходе процесса асептического производства, любая деятельность должна быть сведена к минимуму; передвижения персонала должны подчиняться правилам и контролироваться с целью избежания выделения частиц и микроорганизмов вследствие повышенной активности персонала. Учитывая специфику применяемой технологической одежды, следует обеспечить персоналу комфортные условия по температуре и влажности.
46 Микробное загрязнение сырья и исходных материалов должно быть минимальным. Спецификации на них должны включать в себя требования к микробиологической чистоте, если необходимость этого была установлена в процессе контроля.
47 Использование в чистых зонах тары (упаковок) и материалов, способных выделять волокна, должно быть минимальным.
48 Следует принимать меры по предотвращению загрязнения готового продукта частицами.
49 При работе с различными деталями, тарой и оборудованием после их окончательной очистки и обработки должно быть исключено их повторное загрязнение.
50 Интервалы времени между мойкой, сушкой и стерилизацией упаковки, деталей и оборудования, а также между их стерилизацией и использованием должны быть сведены к минимуму. Длительность этих интервалов должна определяться с учетом условий хранения.
51 Время между началом приготовления раствора и его стерилизацией или стерилизующей фильтрацией должно быть минимальным. Для каждого продукта следует установить максимально допустимое время с учетом состава продукта и установленного порядка хранения.
52 Перед стерилизацией следует проводить отбор проб для проведения микробиологического контроля. Следует установить пределы уровня загрязнения, проверяемого непосредственно перед стерилизацией. Величина этих пределов зависит от эффективности применяемого метода стерилизации. При необходимости проверяют отсутствие пирогенов. Все растворы (особенно инфузионные растворы больших объемов) следует пропускать через фильтры, удерживающие микроорганизмы, устанавливаемые непосредственно перед наполнением.
53 Детали, компоненты первичной упаковки, оборудование и другие предметы, используемые в чистых зонах асептического производства, должны стерилизоваться и поступать в чистую зону через проходные стерилизаторы, герметично встроенные в стены, либо передаваться другим способом, защищающим от внесения загрязнений. Негорючие газы должны проходить через фильтры, задерживающие микроорганизмы.
54 Следует проводить аттестацию (валидацию) любого нового процесса. Результаты аттестации (валидации) должны подтверждаться через определенные интервалы времени, основываясь на опыте работы, или при внесении любых значительных изменений в процесс или оборудование.
Стерилизация
55 Все процессы стерилизации должны быть аттестованы (валидированы). Требуется особое внимание, если применяемый метод стерилизации не описан в действующей Фармакопее или используется для продукта, не являющегося простым водным или масляным раствором. Предпочтительным является метод тепловой стерилизации. В любом случае метод стерилизации должен соответствовать лицензии на производство и регистрационному досье.
56 Для любого процесса стерилизации следует показать (с помощью физических измерений и биологических индикаторов) его пригодность для данной продукции и эффективность для достижения требуемых условий стерилизации во всех частях загрузки каждого вида. Соответствие процесса заданным требованиям следует подтверждать через определенные интервалы времени (не менее чем один раз в год или после любых существенных изменений в оборудовании). Протоколы всех испытаний следует хранить.
57 Процесс стерилизации должен обеспечивать эффективность стерилизации всего объема загрузки.
58 Для всех процессов стерилизации должны быть разработаны и аттестованы (валидированы) схемы загрузки.
59 Биологические индикаторы следует рассматривать в качестве дополнительного метода контроля стерилизации. Биологические индикаторы должны храниться и использоваться согласно инструкциям изготовителя, а их качество должно контролироваться с помощью методов позитивного контроля. При использовании биологических индикаторов следует предусмотреть меры, исключающие возможность микробиологического загрязнения от самих биологических индикаторов.
60 Следует четко определить меры, обеспечивающие разделение продуктов, прошедших и не прошедших стерилизацию. На каждой корзине, лотке или другой емкости для продукта или компонентов должна быть четкая этикетка с названием материала, номером серии и указанием, прошел он стерилизацию или нет. Для обозначения, прошла ли серия (или подсерия) процесс стерилизации или нет, можно использовать такие индикаторы, как автоклавная лента, но они не дают надежного указания на то, что серия действительно стерильна.
61 Для каждого цикла стерилизации следует оформлять протоколы, которые должны утверждаться и входить составной частью в протокол серии готовой продукции.
Тепловая стерилизация
62 Каждый цикл тепловой стерилизации должен записываться на диаграмме "время - температура" в достаточно большом масштабе или регистрироваться с необходимой точностью при помощи другого оборудования. Расположение датчиков температуры, используемых для контроля и/или записи, должно быть определено во время аттестации (валидации) и, по возможности, проверено по другому независимому датчику температуры, расположенному в том же месте.
63 Допускается использовать химические и биологические индикаторы, но они не должны заменять физические измерения.
64 До начала отсчета продолжительности стерилизации должно быть предусмотрено достаточное время, обеспечивающее достижение требуемой температуры всего объема загрузки. Это время должно определяться для каждого вида загрузки отдельно.
65 После завершения высокотемпературной фазы цикла тепловой стерилизации следует принять меры против загрязнения загрузки в период охлаждения. Следует стерилизовать любую охлаждающую жидкость или газ, вступающие в контакт с продуктом, кроме случаев, когда возможность использования негерметичных упаковок исключена и приведены соответствующие доказательства.
Влажное тепло (пар)
66 При стерилизации влажным теплом (паром) следует контролировать температуру и влажность. Как правило, средства управления должны быть независимы от средств контроля и записывающих устройств. Если для этой цели используются автоматические системы управления и контроля, они должны быть аттестованы (валидированы), чтобы гарантировать их соответствие критическим требованиям процесса. Нарушения в ходе процесса должны регистрироваться системой и контролироваться оператором. В ходе процесса стерилизации показания независимого датчика температуры должны постоянно сверяться с данными диаграммы записывающего устройства. Для стерилизаторов, имеющих сток в дне камеры, может возникнуть необходимость регистрации температуры в этой точке в течение всего цикла стерилизации. Если в цикл стерилизации входит фаза вакуумирования, то следует достаточно часто проводить проверки камеры на герметичность.
67 Стерилизуемые предметы, не находящиеся в герметичных упаковках, должны быть завернуты в материал, пропускающий воздух и пар, но предотвращающий повторное загрязнение этих предметов после стерилизации. Необходимо обеспечить контакт всех частей загрузки со стерилизующим агентом при заданных температуре и времени.
68 Для стерилизации должен использоваться пар соответствующего качества, не содержащий включений в таком количестве, при котором может произойти загрязнение продукта или оборудования.
Сухое тепло (жар)
69 При стерилизации сухим теплом (жаром) должны быть предусмотрены циркуляция воздуха внутри камеры и поддержание избыточного давления с целью предотвращения попадания внутрь нестерильного воздуха. Любой поступающий внутрь воздух должен проходить через фильтры высокой эффективности (типа НЕРА). Если стерилизация предусматривает устранение пирогенов, то при аттестации (валидации) следует выполнять тесты на эндотоксины.
Радиационная стерилизация
70 Радиационная стерилизация используется главным образом для стерилизации термочувствительных материалов и продуктов. Многие лекарственные средства и некоторые упаковочные материалы чувствительны к ионизирующему излучению. В связи с этим данный метод допускается только при экспериментальном подтверждении отсутствия его вредного влияния на продукт. Как правило, облучение ультрафиолетовым излучением не является приемлемым методом стерилизации.
71 Во время процесса стерилизации должна измеряться поглощенная доза ионизирующего излучения. Для этого следует использовать дозиметры, показания которых не зависят от используемой мощности дозы излучения, но которые дают количественное значение поглощенной дозы непосредственно в продукции. Для каждой загрузки в стерилизуемой продукции должно находиться достаточное количество дозиметров, расположенных таким образом, чтобы в облучаемой зоне всегда находился дозиметр. Полимерные дозиметры можно применять только в течение установленного срока действия калибровки с измерением их оптической плотности в течение короткого времени после облучения.
72 В качестве средства дополнительного контроля могут использоваться биологические индикаторы.
73 Методики аттестации (валидации) должны гарантировать учет возможных изменений плотности укладки стерилизуемой продукции.
74 При проведении процесса радиационной стерилизации не должно быть смешивания облученной и необлученной продукции. На каждой упаковке следует использовать чувствительные к излучению цветовые индикаторы для того, чтобы различить упаковки, прошедшие и не прошедшие облучение.
75 Суммарная поглощенная доза излучения должна быть набрана в течение времени, отведенного на процесс стерилизации.
Стерилизация оксидом этилена
76 Метод стерилизации оксидом этилена может использоваться только там, где нельзя применять другие методы стерилизации. При проведении аттестации (валидации) следует показать, что процесс стерилизации оксидом этилена не оказывает вредного влияния на продукцию, а заданные условия и длительность дегазации позволяют снизить остаточную концентрацию газа и продуктов реакции до допустимых уровней, определенных для данного вида продукции или материала.
77 Существенное значение имеет непосредственный контакт между газом и микроорганизмами. Следует принять меры предосторожности от включения микроорганизмов в материал (например, в кристаллы или лиофилизированный белок). На процесс стерилизации оксидом этилена могут оказать существенное влияние вид и количество упаковочного материала.
78 Температуру и влажность загрузки перед обработкой газом следует привести в соответствие с требованиями процесса. Требуемое для этого время должно быть по возможности минимальным.
79 Каждый цикл стерилизации должен контролироваться с использованием соответствующих биологических индикаторов, распределенных в достаточном количестве по всему объему загрузки. Полученная информация должна оформляться документально и входить составной частью в протокол на серию готовой продукции.
80 Для каждого цикла стерилизации должны быть оформлены протоколы, содержащие данные о длительности цикла, давлении, температуре и влажности в камере во время процесса, концентрации газа и данные об общем количестве использованного газа. Давление и температура должны регистрироваться в течение всего цикла стерилизации на диаграмме. Эти данные должны входить составной частью в протокол на серию готовой продукции.
81 После стерилизации загрузку следует хранить под контролем в вентилируемых помещениях, чтобы обеспечить снижение остаточной концентрации газа и продуктов реакции до определенного уровня. Этот процесс подлежит аттестации (валидации).
Фильтрация лекарственных средств, которые не могут быть стерилизованы в первичной упаковке
82 Проведение стерилизующей фильтрации не является достаточным условием стерилизации, если возможна стерилизация продукта в окончательной первичной упаковке. Предпочтительным является метод стерилизации паром. Если стерилизация продукта в окончательной первичной упаковке невозможна, то перед наполнением растворов или жидкостей в предварительно стерилизованную первичную упаковку их следует пропускать через стерильные фильтры с номинальным размером пор не более 0,22 мкм или с эквивалентными свойствами по удержанию микроорганизмов. Такие фильтры могут задерживать большую часть бактерий или плесневых грибов, но не все вирусы или микоплазмы. По возможности процесс фильтрации следует дополнять соответствующей термообработкой.
83 В связи с тем, что у метода фильтрации есть дополнительный риск загрязнения микроорганизмами по сравнению с другими способами стерилизации, непосредственно перед наполнением можно рекомендовать повторную фильтрацию продукта через дополнительный удерживающий микроорганизмы стерилизующий фильтр. Окончательную стерилизующую фильтрацию продукта следует проводить как можно ближе к месту наполнения.
84 Следует использовать фильтры с минимальным отделением волокон.
85 Перед использованием стерилизующего фильтра и сразу после его использования следует проверить отсутствие у него повреждений таким методом, как тест на "точку пузырька", методом диффузионного потока или выдержкой под давлением.
При аттестации следует измерять время, необходимое для фильтрации раствора заданного объема, и перепад давления на фильтре. Любые значительные отклонения в процессе производства от аттестованных показателей следует регистрировать и анализировать. Результаты этих проверок должны быть включены в протоколы на серию продукции. Сразу после использования следует подтверждать целостность критических газовых и воздушных фильтров. Целостность других фильтров необходимо подтверждать через соответствующие интервалы времени.
86 Не допускается использовать один и тот же фильтр в течение более одного рабочего дня, за исключением случаев, когда более длительное его использование подтверждено аттестацией (валидацией).
87 Применяемый фильтр не должен оказывать влияние на продукт, задерживая его компоненты или выделяя в него какие-либо посторонние вещества.
Завершение производства стерильной продукции
88 Для укупоривания первичной упаковки следует использовать аттестованные (валидированные) методы. Первичные упаковки, герметизированные запайкой (например, стеклянные или пластмассовые ампулы), подлежат 100%-ной проверке на герметичность. Герметичность других первичных упаковок должна проверяться по соответствующим методикам.
89 Первичные упаковки, герметизированные под вакуумом (вакуумные упаковки), должны проверяться на сохранение вакуума с определенной периодичностью.
90 Первичные упаковки с продукцией для парентерального введения необходимо проверять индивидуально на наличие посторонних включений или других дефектов. Визуальный контроль следует проводить при требуемых уровнях освещенности и фоне рабочего поля. Следует регулярно проверять зрение операторов, выполняющих визуальный контроль (если операторы используют очки, то проверка зрения проводится в очках). В ходе визуального контроля продукции рекомендуется организовывать частые перерывы в работе операторов. При использовании других методов контроля этот процесс необходимо аттестовать (валидировать); состояние оборудования следует периодически проверять. Результаты визуального контроля должны быть оформлены документально.
Контроль качества
91 Испытание готового продукта на стерильность следует рассматривать лишь как завершающий этап в последовательности мер, обеспечивающих стерильность продукта. Тест на стерильность должен быть аттестован (валидирован) для продукции каждого вида.
92 При получении разрешения на выпуск стерильной продукции по параметрам следует уделить особое внимание аттестации (валидации) и контролю всего технологического процесса.
93 Образцы продукции, которые были отобраны для проведения испытания на стерильность, должны быть репрезентативными для всей серии продукции, особенно для тех частей серии, вероятность загрязнения которых максимальна, например:
a) для продуктов, расфасованных в асептических условиях, в отобранные образцы должны входить первичные упаковки, наполненные в начале и в конце серии, и после любого существенного вмешательства в ходе технологического процесса;
b) для продуктов, прошедших тепловую стерилизацию в первичной упаковке, особое внимание следует уделить отбору образцов из потенциально наиболее холодной части загрузки стерилизатора.
Приложение 2
Производство медицинских биологических препаратов
Качество медицинских биологических препаратов и порядок их контроля во многом определяются технологией производства, от которой в значительной степени зависит отнесение медицинских биологических препаратов к тому или иному виду. В данном приложении рассматриваются следующие медицинские биологические препараты:
a) микробные культуры, за исключением полученных по технологии рекомбинантной ДНК;
b) микробные и клеточные культуры, полученные по рекомбинантной ДНК технологии или методом гибридизации;
c) экстракты из биологических тканей;
d) живые агенты, выращенные в эмбрионах или животных.
К таким медицинским биологическим препаратам относятся: вакцины, иммунные сыворотки, антигены, гормоны, цитокины, ферменты и другие продукты ферментации (в т.ч. моноклональные антитела и продукты, получаемые из рекомбинантной ДНК).
К продуктам, указанным в перечислении а), применимы не все требования настоящего приложения.
Примечание - При подготовке настоящего стандарта были учтены общие требования, предъявляемые Всемирной организацией здравоохранения (ВОЗ) к организации производства и контрольным лабораториям.
В настоящем стандарте не приведены требования к специфическим видам биологических препаратов. Требования к ним даны в специальных нормативных документах.
Общие положения
Производство медицинских биологических препаратов имеет свою специфику, определяемую характером продукции и технологией производства. При производстве и контроле медицинской биологической продукции необходимо принимать специальные меры предосторожности.
В отличие от обычных лекарственных средств, которые производятся с использованием химических и физических методов, обеспечивающих высокую степень стабильности, производство медицинских биологических препаратов связано с биологическими процессами и материалами, такими как культивирование клеток или экстракция материала из живых организмов. Эти биологические процессы характеризуются вариабельностью, что приводит к непостоянству спектра и природы сопутствующих продуктов. Более того, материалы, используемые в процессах культивирования, сами по себе являются субстратами для роста микроорганизмов.
Контроль медицинских биологических препаратов включает в себя биологические аналитические методы, которые характеризуются более высокой степенью вариабельности, чем физико-химические аналитические методы. Поэтому при производстве медицинских биологических препаратов особое значение имеют методы внутрипроизводственного контроля.
Персонал
1 Все сотрудники, работающие в зонах производства медицинских биологических препаратов (в т.ч. персонал, занятый очисткой, обслуживанием или контролем качества), должны проходить дополнительное обучение в соответствии с их обязанностями и особенностями производимой продукции. Сотрудники должны иметь достаточную подготовку в области личной гигиены и микробиологии.
2 Для успешного выполнения своих обязанностей лицам, ответственным за производство и контроль качества продукции, наряду с достаточным практическим опытом следует иметь необходимую базовую подготовку по бактериологии, биологии, биометрии, химии, медицине, фармации, фармакологии, вирусологии, иммунологии и ветеринарии.
3 Для обеспечения безопасности производимой продукции следует учитывать иммунный статус сотрудников. Все сотрудники, занятые в производстве, техническом обслуживании, проведении испытаний и уходе за животными (в том числе и контролеры), должны, по мере необходимости, проходить вакцинацию, а также регулярное медицинское обследование. Помимо очевидной необходимости защиты персонала от инфицирующих агентов, сильнодействующих токсинов или аллергенов, следует предотвращать загрязнение серии продукции инфицирующими агентами. Как правило, допуск посторонних лиц в производственные зоны должен быть строго ограничен.
4 Необходимо исключить допуск в производственную зону сотрудников с какими-либо изменениями иммунного статуса, который может отрицательно повлиять на качество производимого продукта. В производстве вакцины БЦЖ и препаратов туберкулина могут быть заняты только сотрудники, которые регулярно проходят проверки иммунного статуса или рентгенографическое обследование грудной клетки.
5 В течение рабочего времени не допускается переход сотрудников из зон, где возможен контакт с живыми микроорганизмами или животными, в зоны работы с другой продукцией или другими микроорганизмами. Если подобных переходов избежать невозможно, персонал, занятый в таком производстве, должен строго выполнять требования по деконтаминации, в т.ч. смену одежды и обуви, и, при необходимости, принимать душ.
Помещения и оборудование
6 Требования к чистоте производственных помещений по микроорганизмам и частицам зависят от вида продукта и технологической стадии, с учетом уровня загрязнения исходных материалов и опасности загрязнения готовой продукции.
7 Ввиду опасности перекрестного загрязнения медицинских биологических препаратов, особенно на стадиях работы с живыми организмами, могут потребоваться дополнительные меры предосторожности, например, использование специально предназначенного оборудования и помещений, организация производства по принципу отдельных циклов работы и использование закрытых систем. Уровень разделения, необходимый для предотвращения перекрестного загрязнения, определяется видом продукта и параметрами используемого оборудования.
8 Для производства вакцины БЦЖ и работы с живыми микроорганизмами, используемыми в производстве препаратов туберкулина, как правило, необходимо применять только специальные помещения и оборудование.
9 При работе с Bacillus anthracis, Clostridium botulinum и Clostridium tetani до завершения процесса их инактивации необходимо использовать только специальные помещения и оборудование.
10 Для других спорообразующих организмов может применяться организация производства по принципу отдельных циклов при условии, что при этом используются помещения и оборудование, специально предназначенные для этой группы продуктов, и одновременно производится продукция только одного вида.
11 Для таких продуктов, как моноклональные антитела и продукты, производимые по рекомбинантной ДНК технологии, допускается их одновременное производство в одной зоне с использованием закрытых систем биоферментаторов.
12 После стадии выращивания последующие технологические стадии могут проводиться для разных продуктов одновременно в одной производственной зоне при условии принятия адекватных мер по предотвращению перекрестного загрязнения. Для убитых вакцин и анатоксинов такое параллельное производство возможно только после инактивации культуры или после ее детоксикации.
13 Для производства стерильной продукции следует использовать помещения с избыточным давлением. Для предотвращения распространения загрязнения за пределы специальных зон, подвергаемых воздействию патогенов, в этих зонах следует создавать отрицательный перепад давления.
Боксы с отрицательным перепадом давления, используемые для работы с патогенами в асептических условиях, следует окружать стерильными зонами с избыточным давлением.
14 При выборе оборудования фильтрации воздуха следует учитывать особенности производственных зон. Как правило, не применяется рециркуляция воздуха из зон, в которых выполняется работа с живыми патогенными микроорганизмами (за исключением обоснованных случаев).
15 Планировка производственных зон и конструкция оборудования должны обеспечивать возможность проведения их эффективной очистки и деконтаминации (например, с использованием метода фумигации). Методики очистки и деконтаминации должны быть аттестованы (валидированы).
16 Конструкция оборудования, используемого для работы с живыми микроорганизмами, должна исключать возможность загрязнения этих культур из внешних источников в ходе технологического процесса.
17 Конструкция трубопроводов, вентилей и фильтров очистки воздуха должна обеспечивать удобство их очистки и стерилизации. Рекомендуется использовать системы "очистка на месте" (clean in place) и "стерилизация на месте" (sterilize in place). Конструкция вентилей на ферментаторах должна предусматривать возможность их стерилизации паром. Фильтры должны быть гидрофобными, срок их службы должен быть определен в ходе аттестации (валидации).
18 Конструкция первичной изоляции должна исключать риск утечки, что следует подтверждать испытаниями.
19 Жидкие отходы, которые могут содержать патогенные микроорганизмы, должны проходить эффективную деконтаминацию.
20 Вследствие вариабельности биологических продуктов или процессов в ходе технологического процесса может возникнуть необходимость в отмеривании или взвешивании каких-либо добавок или ингредиентов (например, буферов), поэтому в производственной зоне допускается хранить небольшие запасы этих веществ.
Виварии и уход за животными
21 Животные используются для производства ряда медицинских биологических препаратов: вакцины против полиомиелита (обезьяны), сыворотки против ядов змей (лошади и козы), вакцины против бешенства (кролики, мыши и хомяки) и сывороточный гонадотропин (лошади). Животные используются также для контроля качества большинства сывороток и вакцин, например, мыши (вакцина против коклюша), морские свинки (БЦЖ вакцина), а также для проведения испытаний на пирогенность (кролики).
22 Общие требования к организации вивариев, уходу за животными и карантину приведены в соответствующей нормативной документации. Виварии, в которых содержатся животные, используемые для производства и контроля качества медицинских биологических препаратов, должны быть отделены от зон производства и контроля качества. Состояние здоровья животных, которые служат источниками исходных материалов или используются для контроля качества продукции и проведения испытаний на безопасность, должно контролироваться, а результаты контроля фиксироваться в документации. Для персонала, занятого в этих зонах, должны быть предусмотрены комнаты для переодевания и соответствующая рабочая одежда. При использовании обезьян для производства или контроля качества медицинских биологических препаратов необходимо учитывать особые требования, указанные в действующих "Нормативах ВОЗ для биологических веществ" N 7.
Документация
23 В спецификации на исходные материалы для производства биологических медицинских препаратов следует включать дополнительную информацию об источнике, происхождении, методе производства и применяемых методах контроля качества, в частности, микробиологического контроля.
24 Как правило, спецификации требуются также для промежуточных и нерасфасованных медицинских биологических препаратов.
Производство
Исходные материалы
25 Следует четко определять источник, происхождение и пригодность исходных материалов для использования. Если проведение необходимых испытаний занимает много времени, допускается начинать обработку исходных материалов до получения результатов этих испытаний. В таких случаях разрешение на выпуск серии готовой продукции зависит от положительных результатов испытаний исходных материалов.
26 При необходимости стерилизации исходных материалов предпочтительным является тепловой метод. Для инактивации биологических материалов могут использоваться другие допустимые методы (например, обработка ионизирующим излучением).
Система посевных материалов и банков клеток
27 Для предотвращения нежелательного изменения свойств, которое может произойти вследствие многократных пересевов или большого числа генераций (пассажей), производство медицинских биологических препаратов, получаемых из культур микроорганизмов, культур клеток или размножением в эмбрионах и животных, должно быть основано на системе главного и рабочего банков посевных культур и/или клеток.
28 Количество генераций (удвоений, пассажей) между посевным материалом или банком клеток и готовой продукцией должно соответствовать требованиям нормативной документации и регистрационного досье. Масштабирование процесса не должно изменять это фундаментальное соотношение.
29 Посевные материалы и банки клеток необходимо соответствующим образом характеризовать и проверять на отсутствие контаминации. В дальнейшем их пригодность должна быть подтверждена стабильностью характеристик и качеством последовательных серий продукции. Посевные материалы и банки клеток следует создавать, хранить и использовать таким образом, чтобы риск их загрязнения или изменения был минимальным.
30 Посевные материалы и банки клеток необходимо создавать в контролируемой среде, обеспечивающей защиту посевных материалов и банков клеток и работающего с ними персонала. При создании посевного материала и банка клеток не допускается одновременная работа в одной зоне или одним и тем же персоналом с другими живыми или инфицирующими материалами (например, вирусами, линиями или штаммами клеток).
31 Данные, подтверждающие стабильность и воспроизводимость посевных материалов и банков клеток, необходимо оформлять в виде документов. Емкости, предназначенные для их хранения, должны быть герметичными, четко маркированными и храниться при необходимой температуре. Необходимо тщательно вести документальный учет хранящихся емкостей. В холодильных установках температуру хранения следует непрерывно регистрировать, а в установках с жидким азотом - контролировать соответствующим образом. Все отклонения параметров хранения за установленные границы и вытекающие из этого корректирующие действия должны быть оформлены протоколом.
32 Работа с посевными материалами и банками клеток выполняется только специально назначенным персоналом под контролем ответственного лица. Доступ к хранящимся посевным материалам и банкам клеток следует контролировать. Различные посевные материалы и банки клеток следует хранить таким образом, чтобы исключалась возможность их подмены или перекрестного загрязнения. Рекомендуется разделять посевные материалы и банки клеток и разные части хранить отдельно во избежание их потери.
33 Операции со всеми емкостями, содержащими главный и рабочий банки клеток и посевных материалов, следует проводить в одном порядке. Емкость, однажды взятая из хранилища, не может быть туда возвращена.
Принципы работы
34 Следует проверять ростовые свойства среды культивирования.
35 Добавление материалов или культур в ферментаторы и другие сосуды, а также отбор проб из них необходимо проводить в тщательно контролируемых условиях, обеспечивающих невозможность внесения загрязнений. При внесении добавок или отборе проб необходимо контролировать правильность подсоединения сосудов.
36 Процессы центрифугирования или смешивания продуктов могут привести к образованию аэрозолей, поэтому во избежание переноса живых микроорганизмов эти процессы следует проводить в изолированных зонах.
37 По возможности, среда культивирования должна быть стерилизована на месте (in situ). При подаче в ферментаторы газов, сред, кислот или щелочей, пеногасителей и т.п., по возможности, следует использовать стерилизующие фильтры, встроенные в линии подачи.
38 Особое внимание необходимо уделять аттестации (валидации) методов удаления или инактивации вирусов (см. соответствующую нормативную документацию).
39 При инактивации или удалении вирусов в ходе производства необходимо принять меры против повторного загрязнения обработанной продукции со стороны необработанной продукции.
40 В хроматографических методах используются разные виды оборудования, которое должно быть специально предназначено для проведения очистки одного типа продукции. Между сериями продукции это оборудование должно проходить стерилизацию или дезинфекцию. Не рекомендуется использовать одно и то же оборудование на разных технологических стадиях. Следует установить критерии приемлемости, срок службы и методы стерилизации или дезинфекции колонок.
Контроль качества
41 В обеспечении стабильности качества медицинских биологических препаратов особое значение имеет внутрипроизводственный контроль. Контрольные операции, имеющие решающее значение для качества продукции (например, отсутствие вирусов), но которые не могут быть проведены на готовой продукции, следует проводить на соответствующих промежуточных стадиях производства.
42 Образцы промежуточных продуктов необходимо хранить в соответствующих условиях, и их количество должно быть достаточным для повторения или подтверждения испытаний по контролю качества серии продукции.
43 Некоторые технологические процессы, например, ферментация, требуют непрерывного контроля параметров. Эти данные должны входить составной частью в протокол на серию продукции.
44 При использовании в производстве перевиваемых культур их специфику следует учитывать в требованиях к контролю качества.
Приложение 3
Производство радиофармацевтических препаратов
Общие положения
Производство радиофармацевтических препаратов и работа с ними представляют потенциальную опасность. Степень риска зависит, в частности, от вида излучения и периода полураспада радиоактивных изотопов. Особое внимание следует уделять предотвращению перекрестного загрязнения, распространению радиоактивного загрязнения и утилизации радиоактивных отходов. Нужно учитывать, что радиофармацевтические препараты часто изготавливаются небольшими сериями. Вследствие того, что период полураспада многих радиофармацевтических препаратов невелик, их реализация может осуществляться до завершения испытаний по контролю качества. В этом случае особое значение приобретает непрерывная оценка эффективности системы обеспечения качества.
Примечание - Производство радиофармацевтических препаратов должно соответствовать требованиям нормативных документов, содержащих основные требования к защите населения и персонала от воздействия ионизирующего излучения, а также требованиям национального законодательства.
Персонал
1 Все сотрудники (в т.ч. персонал, занятый уборкой и обслуживанием), работающие в зонах производства радиоактивной продукции, должны пройти дополнительное обучение и необходимую подготовку по защите от радиации.
Помещения и оборудование
2 Радиоактивную продукцию следует хранить, обрабатывать, упаковывать и контролировать в специально предназначенных и изолированных помещениях. Технологическое оборудование для производства радиофармацевтических препаратов не должно использоваться для других целей.
3 Для предотвращения распространения радиоактивных частиц в зонах, в которых проводятся работы с радиоактивной продукцией, может оказаться необходимым создавать отрицательный перепад давления по отношению к окружающим помещениям. При этом также необходимо предотвращать загрязнение продукции частицами из воздуха рабочей зоны.
4 При производстве стерильной продукции условия в рабочих зонах, в которых продукция или первичная упаковка подвергаются воздействию окружающей среды, должны соответствовать требованиям приложения 1. Эти условия могут обеспечиваться использованием оборудования с однонаправленным (ламинарным) потоком воздуха, подаваемым от НЕРА-фильтров, и воздушными шлюзами. Таким требованиям может удовлетворять полностью изолированное оборудование. Это оборудование должно находиться в чистых зонах, по крайней мере, не ниже D.
5 Как правило, не применяется регенерация воздуха из зон работы (обращения) с радиоактивными веществами (за исключением обоснованных случаев). Для исключения возможности загрязнения окружающей среды радиоактивными веществами необходимо предусматривать соответствующую очистку вытяжного воздуха, а также защиту от попадания воздуха в чистую зону через вытяжные воздуховоды (например, при выключенном вытяжном вентиляторе).
Производство
6 Во избежание перекрестного загрязнения или перепутывания не допускается производство различных типов радиоактивной продукции на одних и тех же установках в одно и то же время.
7 При необходимости принятия решения о выпуске или отбраковке серии продукции до завершения всех необходимых испытаний особое значение имеет проведение аттестации (валидации) процессов, внутрипроизводственного контроля и мониторинга технологических параметров и окружающей среды.
Контроль качества
8 При выпуске серии продукции до завершения всех необходимых испытаний может потребоваться принятие Уполномоченным лицом решения о пригодности серии, оформленного в установленном порядке. Порядок действий в этом случае должен быть изложен в инструкции, содержащей данные о производстве и контроле качества, которые необходимо учесть при принятии решения о пригодности серии продукции. В этой инструкции также должны быть указаны действия Уполномоченного лица в случае, если после реализации серии продукции было выявлено ее несоответствие установленным требованиям.
9 Необходимо хранить образцы каждой серии продукции, если другие указания в документации отсутствуют.
Реализация и отзыв продукции
10 Следует хранить подробную документацию на реализацию продукции. Меры, принимаемые для прекращения использования бракованных радиофармацевтических препаратов, приводятся в инструкциях, которые должны предусматривать отзыв такой продукции в самое короткое время.
Приложение 4
Производство лекарственных средств для животных
(кроме иммунных препаратов)
Настоящее приложение относится ко всем лекарственным средствам для животных, за исключением иммунных препаратов (производство которых рассматривается в приложении 5 к настоящему стандарту) и лекарственных кормов.
Производство добавок к лекарственным кормам
В настоящем приложении использованы следующие термины:
лекарственные корма (medicated feeding stuff): Любая смесь лекарственных средств для животных и кормов, выпускаемая в готовом виде, предназначенная для кормления животных без дальнейшей обработки, которая рассматривается как лекарственное средство благодаря своим лечебным или профилактическим свойствам.
добавки (премиксы) (рге-mix) к лекарственным кормам: Любые лекарственные средства для животных, приготовленные заранее для последующего использования в производстве лекарственных кормов.
1 Производство добавок к лекарственным кормам требует использования большого количества растительных материалов, привлекающих насекомых и грызунов. Помещения для производства добавок следует проектировать, оборудовать и эксплуатировать таким образом, чтобы свести к минимуму опасность проникания насекомых и грызунов (3.4). С целью борьбы с ними следует проводить регулярную обработку помещений.
2 Из-за большого объема пыли, образующегося при производстве нерасфасованных веществ для добавок, особое внимание необходимо уделять мерам по облегчению очистки помещений и предотвращению перекрестного загрязнения (3.14), например, применением герметичных транспортеров и пылепоглотителей. Наряду с использованием такого оборудования следует проводить тщательную и регулярную очистку производственных помещений.
3 Следует обеспечить однородность выполнения стадий технологического процесса, которые оказывают существенное отрицательное влияние на стабильность активных веществ (например, использование пара при производстве гранул).
4 Производство добавок целесообразно организовывать в специализированных зонах, вынесенных за пределы основных производственных помещений. В противном случае эти специализированные помещения следует окружить буферными зонами, чтобы опасность загрязнения других производственных зон была минимальной.
Производство препаратов против эктопаразитов
5 Несмотря на ограничение, указанное в 3.6 настоящего стандарта, препараты против эктопаразитов, предназначенные для наружного применения, относящиеся к лекарственным средствам для животных и включенные в лицензию на производство, могут производиться и наполняться в зонах, предназначенных для изготовления пестицидов, по принципу разделенных во времени циклов производства. Такие зоны не предназначены для производства других видов лекарственных средств для животных.
6 Для предотвращения перекрестного загрязнения следует использовать аттестованные (валидированные) методики очистки. Следует принять меры по обеспечению безопасного хранения препаратов для животных в соответствии с требованиями настоящего стандарта.
Производство лекарственных средств для животных, содержащих пенициллины
7 Использование пенициллинов в ветеринарии не представляет такого риска с точки зрения гиперсенсибилизации животных, как в случае их использования человеком. Зарегистрированные случаи гиперсенсибилизации у лошадей и собак были обусловлены другими токсичными веществами (например, ионофорными антибиотиками для лошадей). Такие препараты рекомендуется производить в специально предназначенных изолированных помещениях (3.6), но эта рекомендация может не распространяться на случаи, когда помещения предназначены только для производства лекарственных средств для животных. Однако необходимо принять все необходимые меры по предотвращению перекрестного загрязнения и обеспечению безопасности персонала в соответствии с требованиями настоящего стандарта. При использовании общих помещений производство продукции, содержащей пенициллины, должно быть организовано по принципам разделенных во времени циклов производства и должно сопровождаться аттестованными (валидированными) методиками деконтаминации и очистки.
Хранение образцов (1.4, перечисления VIII и 6.14)
8 В связи с большими объемами окончательных упаковок некоторых лекарственных средств для животных (в частности, добавок) производителю может оказаться неудобным хранить образцы каждой серии продукции в их окончательной упаковке. Однако производитель должен обеспечить хранение достаточного количества архивных образцов каждой серии продукции в соответствии с требованиями настоящего стандарта.
9 В любых случаях упаковка для хранения архивных образцов должна быть изготовлена из того же материала, что и маркированная первичная упаковка, в которой этот продукт реализуется на рынке.
Стерильные лекарственные средства для животных
10 Лекарственные средства для животных, подлежащие финишной стерилизации, могут производиться в чистых зонах более низкого класса, чем это требуется в приложении 1 (если решение об этом принято в установленном порядке), однако тип зоны помещений должен быть не ниже D.
Приложение 5
Производство иммунных лекарственных средств для животных
Общие положения
Производство иммунных лекарственных средств для животных имеет ряд особенностей, которые следует принимать во внимание при организации системы обеспечения качества.
Из-за большого количества видов животных и сопутствующих патогенных микроорганизмов спектр продукции, производимой на предприятии, может быть очень широким при небольшом объеме производства. В связи с этим производство обычно организуется по принципу отдельных циклов. Более того, ввиду особенностей этого производства (стадии культивирования, отсутствие финишной стерилизации и т.п.) необходима тщательная защита продукции от прямого и перекрестного загрязнений. Большое внимание следует уделять защите окружающей среды, особенно при использовании патогенных или экзотических микроорганизмов. При использовании микроорганизмов, патогенных для человека, особенно тщательно следует защищать работающий персонал.
С учетом этих факторов, а также нестабильности свойств, характерной для иммунных препаратов, и относительно низкой эффективности проводимых испытаний (в частности, при контроле качества готовой продукции), система обеспечения качества приобретает особенно важное значение. При производстве иммунных лекарственных средств для животных особую роль играет постоянный контроль за соблюдением всех требований, изложенных в основной части стандарта и настоящем приложении. Следует проводить непрерывную оценку данных, получаемых в ходе контроля различных аспектов производства (оборудование, помещения, продукция и т.п.) на соответствие требованиям настоящего стандарта. Решения и действия, предпринятые на основании этой оценки, должны быть оформлены документально.
Персонал
1 Все сотрудники (включая персонал, занятый очисткой и обслуживанием), работающие в зонах производства иммунной продукции, должны пройти обучение по личной гигиене и микробиологии и дополнительное обучение в соответствии со спецификой производимой продукции.
2 Лица, ответственные за производство и контроль качества, должны иметь базовую подготовку по следующим предметам (всем или некоторым): бактериологии, биологии, биометрии, химии, иммунологии, медицине, паразитологии, фармации, фармакологии, вирусологии и ветеринарии, а также необходимые знания по защите окружающей среды.
3 Персонал должен быть защищен от возможного заражения микроорганизмами, используемыми в производстве. При использовании микроорганизмов, известных как возбудители болезней человека, необходимо принимать особые меры по защите персонала, работающего с этими микроорганизмами или экспериментальными животными.
В случае необходимости персонал должен прививаться соответствующими вакцинами и проходить медицинское обследование.
4 Необходимо принимать соответствующие меры против переноса микроорганизмов людьми за пределы производства. В зависимости от вида микроорганизмов к таким мерам относятся полное переодевание и обязательное принятие душа перед выходом из производственной зоны.
5 При производстве иммунной продукции особую важность представляет риск прямого и перекрестного загрязнений, вызываемых персоналом.
Для предотвращения прямого загрязнения, вызываемого персоналом, предусматривается выполнение ряда мер и инструкций, включающих в себя использование защитной одежды на различных стадиях технологического процесса.
Для предотвращения перекрестного загрязнения персонал не должен перемещаться из одной зоны в другую без соблюдения правил, исключающих риск переноса загрязнения. Эти правила должны быть изложены в инструкции. В течение рабочего времени персонал не должен переходить из зон, где возможно загрязнение живыми микроорганизмами или содержатся животные, в помещения, где работают с другими продуктами или микроорганизмами. Если таких перемещений избежать невозможно, персонал, занятый в таком производстве, должен следовать четко установленным методикам деконтаминации, в т.ч. выполнять смену одежды, обуви, и, по мере необходимости, принимать душ.
Принято считать, что персонал не сталкивается с риском загрязнения при входе в изолированную зону для проверки культур, находящихся в герметически закрытых контейнерах, поверхность которых прошла деконтаминацию, если в этой зоне в течение последних 12 ч не велись работы с открытыми микроорганизмами (за исключением случаев экзотических микроорганизмов).
Помещения
6 При проектировании помещений должна быть предусмотрена защита как продукции, так и окружающей среды. Это может быть достигнуто за счет использования изолированных, чистых, чистых/изолированных или контролируемых зон.
7 Операции с живыми микроорганизмами должны проводиться в изолированных зонах. Уровень изоляции должен зависеть от патогенности микроорганизмов и от того, были ли они классифицированы как экзотические.
8 Операции с инактивированными микроорганизмами следует проводить в чистых зонах. Чистые зоны следует также использовать при работе с неинфицированными клетками, выделенными из многоклеточных организмов, и в некоторых случаях при работе со средами, прошедшими стерилизующую фильтрацию.
9 Операции с открытыми продуктами или компонентами первичной упаковки, которые не подлежат дальнейшей стерилизации, следует проводить в боксе (установке) с однонаправленным (ламинарным) потоком воздуха (зона А), находящемся в зоне В.
10 Если производство находится в том же здании, то другие операции с живыми микроорганизмами (контроль качества, исследования и диагностика и т.п.) должны выполняться в изолированных и выделенных помещениях. Степень изоляции зависит от патогенности этого вида микроорганизмов и от того, был ли он классифицирован как экзотический. При выполнении диагностических операций существует риск загрязнения высокопатогенными микроорганизмами. Уровень изоляции должен соответствовать всем вышеперечисленным рискам. Изоляция может потребоваться и в случаях, когда контроль качества и другие операции проводятся в зданиях, расположенных вблизи производственных зданий.
11 Изолированные помещения должны легко дезинфицироваться. При проектировании этих помещений следует предусматривать:
a) отсутствие прямого выхода вентилируемого воздуха наружу;
b) наличие вентиляции с обеспечением отрицательного перепада давления (разрежения). Воздух должен удаляться через НЕРА-фильтры. Не допускается рециркуляция воздуха в ту же зону, за исключением случаев его дополнительной очистки НЕРА-фильтрами (обычно это условие выполняется при прохождении рециркулирующего воздуха через приточные НЕРА-фильтры, которые используются для этой зоны). Поступление воздуха из одной зоны в другую допускается в том случае, если вытяжной воздух проходит через два последовательно установленных НЕРА-фильтра. При этом должны быть предусмотрены непрерывный контроль целостности первого фильтра и средства безопасного удаления вытяжного воздуха в случае повреждения этого фильтра;
c) воздух, выходящий из производственных зон, в которых проводится работа с экзотическими микроорганизмами, должен проходить через два последовательно установленных НЕРА-фильтра. При этом рециркуляция воздуха между производственными зонами не допускается;
d) наличие системы сбора и дезинфекции жидких отходов, включая контаминированный конденсат из стерилизаторов, биореакторов и т.п. Твердые отходы, в т.ч. трупы животных, следует дезинфицировать, стерилизовать или сжигать. Контаминированные фильтры необходимо удалять безопасным способом;
e) комнаты переодевания следует проектировать и использовать как воздушные шлюзы и оборудовать, при необходимости, умывальниками и душевыми. Схема перепада давления воздуха должна предотвращать движение воздуха между рабочей зоной и окружающей средой, а также риск загрязнения одежды, используемой вне рабочей зоны;
f) для перемещения оборудования должна быть предусмотрена система воздушных шлюзов, конструкция которой предотвращала бы движение загрязненного воздуха рабочей зоны во внешнюю среду и риск загрязнения оборудования внутри шлюза. Размер шлюза должен позволять проводить эффективную деконтаминацию поверхности перемещаемых материалов. Рекомендуется устанавливать на двери шлюза таймер, позволяющий контролировать время, достаточное для проведения эффективной деконтаминации;
g) во многих случаях для безопасного удаления отходов и подачи стерильных предметов необходим проходной автоклав с двумя дверями.
12 Воздушные шлюзы для передачи материалов и комнаты переодевания следует оборудовать блокирующими устройствами, препятствующими одновременному открыванию более одной двери. В комнаты переодевания следует подавать очищенный воздух того же качества, что и для производственных зон. Они должны быть оборудованы системами удаления воздуха, обеспечивающими воздухообмен, независимый от производственных зон. Воздушные шлюзы для передачи материалов должны, как правило, вентилироваться таким же образом, однако допускается применять невентилируемые шлюзы или шлюзы, оборудованные только приточными системами.
13 Стадии технологического процесса, при которых может произойти загрязнение продукции (хранение клеток, подготовка сред, культивирование вирусов и т.п.), должны выполняться в отдельных зонах. Особая предосторожность требуется при работе с животными и продуктами животного происхождения.
14 Операции с микроорганизмами, проявляющими высокую устойчивость к дезинфекции (например, спорообразующие бактерии), до их инактивации следует выделять в изолированных зонах, специально предназначенных для проведения таких операций.
15 В производственной зоне следует одновременно проводить работы только с микроорганизмами одного вида, за исключением процессов смешения и последующего наполнения.
16 Следует предусматривать возможность проведения дезинфекции производственных зон в промежутках между производственными циклами с использованием аттестованных (валидированных) методов.
17 Допускается производство микроорганизмов в контролируемых зонах при условии, что оно осуществляется в полностью закрытом и стерилизуемом тепловым методом оборудовании, а все соединения после завершения работы и перед разборкой также подвергаются тепловой стерилизации. Допускается сборка соединений под локальным однонаправленным (ламинарным) потоком воздуха при условии, что их количество ограничено, используются соответствующие асептические методы и отсутствует опасность утечки. Параметры процесса стерилизации, используемого перед разборкой соединений, должны пройти аттестацию (валидацию) для всех используемых микроорганизмов. В пределах одной зоны разные продукты могут быть загружены в разные биореакторы только при отсутствии риска случайного перекрестного загрязнения. Работа с микроорганизмами, к которым предъявляются особые требования изоляции, должна выполняться только в специально выделенных зонах.
18 В помещениях для содержания животных, предназначенных для использования (или используемых) в производстве, следует поддерживать режим изолированной и/или чистой зоны и отделять их от помещений для содержания других животных. Помещения, где содержатся животные, используемые для контроля качества продукции (в т.ч. и с применением патогенных микроорганизмов), должны быть изолированы.
19 Доступ в производственные зоны может иметь только персонал, имеющий на это разрешение. Режим доступа в производственные зоны должен быть определен специальной инструкцией.
20 Техническая документация предприятия должна содержать необходимые данные о производственных помещениях.
Эти данные должны включать в себя планы помещений, их экспликацию, пояснительную записку и другие материалы, входящие в проект производства, позволяющие установить назначение и условия использования всех помещений, а также виды биологических агентов, с которыми проводится работа. Потоки людей и продукции также должны быть четко обозначены. Необходимо указать виды животных, содержащихся в вивариях или других помещениях, а также виды работ, выполняемых вблизи производственного участка.
Документация на изолированные и/или чистые зоны должна содержать принципиальную схему вентиляции с указанием мест притока и вытяжки воздуха, фильтров и их спецификаций, кратностей воздухообмена и перепадов давления. Следует указать, между какими помещениями предусмотрен контроль перепада давления с помощью датчиков (индикаторов).
Оборудование
21 Конструкция и монтаж оборудования должны соответствовать специфическим требованиям для каждого вида продукции.
Перед вводом в эксплуатацию оборудования его следует аттестовать, а затем организовать его техническое обслуживание и проведение повторной аттестации.
22 Оборудование должно обеспечивать удовлетворительную первичную изоляцию биологических агентов. Конструкция и монтаж оборудования должны, при необходимости, обеспечивать его легкую и эффективную деконтаминацию и/или стерилизацию.
23 Конструкция и монтаж закрытого оборудования, используемого для первичной изоляции биологических агентов, должны предусматривать предотвращение утечек или формирование капель и аэрозолей.
Вводы и выводы газов должны быть защищены так, чтобы обеспечивать нужную степень изоляции, например, путем использования стерилизующих гидрофобных фильтров.
Для подачи или удаления материалов следует использовать стерилизуемую закрытую систему, либо условия ламинарного потока воздуха.
24 Перед использованием оборудование следует стерилизовать предпочтительно сухим паром под давлением. Другие методы применимы в том случае, если оборудование не допускает стерилизацию паром. Важно не пропустить при обработке такие виды оборудования, как центрифуги и водяные бани.
Оборудование, используемое для очистки, разделения или концентрирования, должно, как минимум, стерилизоваться или дезинфицироваться при переходе от одного вида продукта к другому. Необходимо исследовать влияние методов стерилизации на эффективность и аттестационный статус оборудования с целью определения срока его эксплуатации.
Все методики стерилизации должны быть аттестованы (валидированы).
25 Конструкция оборудования должна исключать возможность перепутывания различных организмов или продуктов. Трубопроводы, клапаны и фильтры должны маркироваться в соответствии с их назначением.
Для упаковок с инфекционными и неинфекционными материалами, а также, как правило, для различных организмов и клеток следует использовать отдельные инкубаторы. Содержание в одном инкубаторе более одного типа организмов или типа клеток допускается только в случае принятия необходимых мер по герметизации, поверхностной деконтаминации и разделению упаковок. Сосуды с культурами должны быть промаркированы индивидуально. Очистка и дезинфекция этих сосудов и инкубаторов могут быть затруднены и требовать особого внимания.
Конструкция и порядок эксплуатации оборудования, используемого для хранения биологических агентов или продуктов, должны исключать любое возможное перепутывание.
Все хранящиеся образцы должны иметь четкую и однозначную маркировку и находиться в контейнерах, защищенных от утечки. Резервные посевные культуры клеток и организмов должны храниться в специально предназначенном оборудовании.
26 Некоторые виды оборудования (например, оборудование, требующее контроля температуры) должны быть оснащены регистрирующими устройствами и/или системами сигнализации. Во избежание отказов, совместно с анализом регистрируемых данных, следует организовать систему профилактического обслуживания.
27 Загрузка лиофильных сушильных установок должна происходить в чистой/изолированной зоне. Операции по разгрузке лиофильной сушильной установки приводят к загрязнению окружающей среды. В связи с этим при использовании лиофильных сушильных установок, открывающихся с одной стороны, чистое помещение должно пройти деконтаминацию до перемещения в эту зону другой серии продукции, за исключением серий с одинаковыми организмами. Двусторонние лиофильные сушильные установки должны стерилизоваться после каждого цикла производства, если только они не открываются в чистую зону.
Стерилизация лиофильных сушильных установок должна производиться в соответствии с пунктом 24 данного приложения. В случае организации работ по принципу отдельных циклов производствае их следует стерилизовать, по крайней мере, после каждого цикла.
Животные и помещения для их содержания (виварии)
28 Общие требования к содержанию животных, помещениям для них, уходу и карантину приведены в соответствующей нормативной документации.
29 Виварии должны быть изолированы от других производственных помещений. Их проекты должны быть выполнены с соблюдением необходимых требований.
30 Санитарное состояние животных, используемых в производстве, необходимо оценивать, контролировать и регистрировать. Правила обращения с некоторыми видами животных приведены в специальных руководствах.
31 Животные, биологические агенты и проводимые испытания должны быть четко маркированы (документированы) в соответствии с установленной системой во избежание риска перепутывания и с целью контроля всех возможных видов опасностей.
Дезинфекция. Уничтожение отходов
32 Дезинфекция и/или уничтожение твердых и жидких отходов могут иметь особенно важную роль при производстве иммунной продукции. В связи с этим необходим тщательный подход к методам и оборудованию, используемым для предотвращения загрязнения окружающей среды, а также к их аттестации (валидации).
Производство
33 Из-за широкого разнообразия продукции, большого количества стадий при производстве иммунных лекарственных средств для животных и характера биологических процессов особое внимание следует уделять строгому соблюдению аттестованных (валидированных) методов, постоянному контролю всех технологических стадий производства и проведению внутрипроизводственного контроля. Особое внимание следует обратить на исходные материалы, среды культивирования и посевной материал.
Исходные материалы
34 Требования к исходным материалам должны быть четко определены в документально оформленных спецификациях. Спецификации должны содержать данные о поставщике, методе производства, его месте расположения и видах животных, из которых извлекаются эти исходные материалы, а также способы их контроля. Особенно важны микробиологические методы контроля.
35 Результаты испытаний исходных материалов должны соответствовать спецификациям. Если проведение испытаний занимает много времени (например, яйца (эмбрионы) безлейкозных линий птиц), может возникнуть необходимость обработки исходных материалов до получения результатов аналитических испытаний. В этих случаях получение разрешения на реализацию готовой продукции зависит от положительных результатов испытания исходных материалов.
36 Особое внимание следует уделять данным о системе обеспечения качества, принятой у поставщика, об оценке пригодности источников материалов и требуемых для обеспечения качества тестах.
37 Там, где это возможно, следует стерилизовать исходные материалы тепловым методом. Допускается использовать и другие аттестованные методы, например, ионизирующее излучение.
Среды
38 Способность сред поддерживать необходимый рост клеток должна быть заранее подтверждена соответствующим образом.
39 Предпочтительно, чтобы среды стерилизовались на месте (in situ) или на линии. Предпочтительным является метод тепловой обработки. Газы, среды, кислоты, щелочи, пеногасители и другие вещества, вводимые в стерильный биореактор, должны, в свою очередь, быть стерильными.
Система посевных материалов и банков клеток
40 Для предотвращения нежелательного изменения свойств посевного материала вследствие многократных пересевов или большого числа генераций, производство иммунных лекарственных средств для животных, получаемых из микробных, клеточных или тканевых культур, или размножением в эмбрионах и животных, должно быть основано на системе посевных материалов или банков клеток.
41 Количество генераций (удвоений, пассажей) между посевным материалом или банком клеток и готовой продукцией должно соответствовать данным нормативной документации и регистрационного досье.
42 Посевные материалы и банки клеток следует соответствующим образом характеризовать и контролировать на наличие загрязнений. Посевные материалы и банки клеток должны создаваться, содержаться и использоваться таким образом, чтобы снизить до минимума риск загрязнения или любого изменения. При создании посевных материалов или банков клеток не допускается одновременная работа в той же зоне или тем же персоналом с другими живыми или инфекционными материалами (например, вирусами или клеточными линиями).
43 Для защиты самого посевного материала или банка клеток, а также работающего с ними персонала и окружающей среды посевные материалы или банк клеток должны создаваться в необходимых условиях.
44 Происхождение, форма и условия хранения посевного материала должны быть полностью описаны. Необходимо иметь доказательства стабильности и воспроизводимости посевных материалов и клеток. Емкости для хранения должны быть герметично закрыты, четко маркированы и храниться при требуемой температуре. Условия хранения должны контролироваться. Необходимо вести тщательный учет каждой единицы хранения.
45 К работе с материалом допускаются только специально назначенные сотрудники. Работа должна проводиться под контролем ответственного лица. Различные посевные материалы и банки клеток следует хранить таким образом, чтобы не допускать перепутывания или перекрестного загрязнения. Желательно разделять посевные материалы и банки клеток на части и хранить их в различных местах, чтобы уменьшить опасность полной потери.
Принципы работы
46 В ходе технологических процессов следует избегать или сводить к минимуму образование капель или пены. Процессы центрифугирования или смешения, которые могут привести к образованию капель, должны проводиться в изолированных или чистых/изолированных зонах во избежание переноса живых организмов.
47 Случайные проливы жидкостей, особенно содержащих живые организмы, необходимо быстро и безопасным способом ликвидировать. Для каждого типа микроорганизмов следует иметь аттестованные (валидированные) методики деконтаминации. Там, где используются различные штаммы бактерий одного вида или очень похожие вирусы, этот процесс может быть аттестован (валидирован) только для одного из них, если данные о существенном различии их устойчивости к воздействию используемого агента отсутствуют.
48 Операции, включающие в себя перемещение таких материалов, как стерильные среды, культуры или продукты, должны проводиться в предварительно стерилизованных закрытых системах. Там, где это невозможно, операции по перемещению материалов должны проводиться в однонаправленном (ламинарном) потоке воздуха.
49 Добавление сред или культур в биореакторы (ферментаторы) и другие сосуды должно проводиться в тщательно контролируемых условиях, обеспечивающих невозможность внесения загрязнения. При добавлении культур необходимо тщательно проверять правильность соединения сосудов.
50 Там, где это необходимо (например, когда два и более ферментатора расположены в одной зоне), отверстия для отбора проб и внесения добавок должны стерилизоваться паром (после подсоединения, перед подачей продукта и перед отсоединением). В других случаях допускается химическая дезинфекция входных отверстий и защита соединений под ламинарным потоком воздуха.
51 Оборудование, лабораторная посуда, внешние поверхности упаковок с продукцией и другие подобные материалы перед перемещением из изолированной зоны должны быть дезинфицированы с использованием аттестованного (валидированного) метода (пункт 47 данного приложения). Особую проблему может вызывать ведение документации на серию продукции. Только абсолютный минимум документации, необходимой для соблюдения требований настоящего стандарта, должен поступать в зону и покидать ее. При случайном загрязнении (например, каплями или аэрозолями) или при использовании экзотических микроорганизмов бумажная документация должна дезинфицироваться в шлюзе для перемещения оборудования или передаваться с использованием фотокопии или факса.
52 Жидкие или твердые отходы, такие как дебрис, после отбора материала из яиц, одноразовые бутыли для культур, нежелательные культуры или биологические агенты целесообразно стерилизовать или дезинфицировать перед удалением их из изолированной зоны. В некоторых случаях приемлемыми считаются и другие методы, например использование герметичных контейнеров или трубопроводов.
53 Необходимо тщательно контролировать, чтобы в производственную зону попадали только те предметы и материалы, в т.ч. документация, которые относятся к производимой продукции. Для контроля за соответствием количества вносимых и выносимых предметов и материалов, во избежание их накопления в производственном помещении, должна существовать система учета.
54 Термостойкие предметы и материалы должны поступать в чистую или чистую изолированную зону через проходные автоклавы или печи. Чувствительные к нагреву предметы и материалы должны поступать через воздушный шлюз с блокируемыми дверями, где эти предметы и материалы подвергаются дезинфекции. Допускается стерилизация предметов и материалов в другом месте, если они поступают через шлюз в двойной оболочке с соблюдением необходимых предосторожностей.
55 Во избежание загрязнения или подмены культуры клеток или микроорганизмов в инкубационный период необходимо принимать меры предосторожности. Должна существовать методика очистки и дезинфекции инкубаторов. Упаковки, находящиеся в инкубаторах, должны быть четко и тщательно маркированы.
56 В одном помещении допускаются одновременные операции только с одним биологическим агентом, за исключением операций по смешению и последующему наполнению или при использовании полностью закрытых систем. В промежутках между операциями с различными живыми биологическими агентами производственные помещения должны проходить эффективную дезинфекцию.
57 Продукты должны инактивироваться путем добавления инактиватора с последующим тщательным перемешиванием. После этого смесь должна переноситься во второй стерильный сосуд, за исключением случаев, когда форма и размер сосуда позволяют его легко переворачивать и встряхивать таким образом, чтобы все внутренние поверхности смачивались конечной смесью культура-инактиватор.
58 Сосуды, содержащие инактивированный продукт, нельзя открывать. Из этих сосудов нельзя отбирать пробы в зонах, где содержатся живые биологические агенты. Всю последующую обработку инактивированных продуктов следует проводить в чистых зонах А и В или в закрытом оборудовании, предназначенном для инактивированных продуктов.
59 Необходимо уделять серьезное внимание аттестации (валидации) методов стерилизации, дезинфекции, удаления вирусов и инактивации.
60 Наполнение, по возможности, следует проводить немедленно после завершения производственных операций. До начала операции наполнения емкости с нерасфасованной продукцией должны быть герметично закрыты, маркированы и храниться при надлежащих температурных условиях.
61 Должна существовать система, обеспечивающая контроль целостности и герметичности упаковок после наполнения.
62 Флаконы, содержащие живые биологические агенты, закрываются крышками таким образом, чтобы обеспечивалась невозможность загрязнения другой продукции или проникание живых агентов в другие зоны или во внешнюю среду.
63 Между наполнением первичных упаковок, их маркировкой и упаковкой по различным причинам может пройти определенный отрезок времени. Должны быть разработаны и документально оформлены методики, описывающие порядок хранения немаркированных контейнеров, обеспечивающие невозможность их подмены и надлежащие условия хранения. Особое внимание следует уделять хранению термо- и светочувствительной продукции. Температура хранения должна быть задана.
64 На каждой технологической стадии следует проводить сопоставление реального и ожидаемого выхода продукции. Все существенные отклонения должны быть расследованы.
Контроль качества
65 В обеспечении стабильности качества биологических лекарственных средств особое значение имеет внутрипроизводственный контроль. Виды контроля, которые имеют решающее значение для оценки качества (например, контроль на отсутствие вирусов), но не могут быть проведены на готовой продукции, должны выполняться на одной из предшествующих стадий производства.
66 Для повторения или подтверждения результатов контроля качества серии продукции может возникнуть необходимость хранения при соответствующих условиях достаточного объема образцов промежуточных продуктов.
67 Может возникнуть необходимость непрерывного контроля параметров в ходе процесса производства (например, непрерывного контроля физических параметров в ходе ферментации).
68 Распространенной практикой является непрерывное культивирование биологической продукции. При таком методе производства необходимо принять во внимание особые требования к организации контроля качества продукции.
Приложение 6
Производство медицинских газов
1 Общие положения
В настоящем приложении рассматривается промышленное производство медицинских газов, которое относится к специализированным промышленным процессам и осуществляется, как правило, на нефармацевтических предприятиях. Приложение не распространяется на производство и обращение медицинских газов в больницах, регулируемое специальными нормами. Тем не менее, соответствующие разделы данного приложения могут быть использованы в качестве основы для организации этой работы.
Производство медицинских газов, как правило, осуществляется в закрытом оборудовании. Благодаря этому риск загрязнения данной продукции из окружающей среды является минимальным. Однако существует риск перекрестного загрязнения другими газами.
Производство медицинских газов следует осуществлять в соответствии с настоящим стандартом, фармакопейными требованиями и другими документами.
Примечание - Требования настоящего приложения распространяются только на организацию производства и контроля качества медицинских газов. Требования промышленной безопасности при их производстве приведены в других нормативных документах.
2 Персонал
2.1 Уполномоченное лицо, ответственное за выдачу разрешений на выпуск медицинских газов, должно иметь специальные знания в области производства и контроля качества медицинских газов.
2.2 Персонал, занятый в производстве медицинских газов, должен понимать требования настоящего стандарта в отношении медицинских газов, знать критически важные аспекты и возможную опасность для пациентов, которую могут представлять лекарственные препараты в форме медицинских газов.
3 Помещения и оборудование
3.1 Помещения
3.1.1 Операции наполнения медицинскими и немедицинскими газами следует осуществлять в зонах, отделенных от зон наполнения немедицинскими газами. Запрещается обмен баллонами (контейнерами) между этими зонами. В исключительных случаях допускается наполнение медицинских и немедицинских газов в одной и той же зоне по принципу отдельных циклов производства при условии, что предусмотрены специальные меры предосторожности и проводится необходимая аттестация (валидация).
3.1.2 Помещения, в которых выполняются операции по производству, проведению испытаний и хранению медицинских газов, должны иметь достаточную площадь для исключения перепутывания. Помещения должны содержаться в чистоте и обеспечивать требуемый порядок работы и хранения продукции.
3.1.3 Размеры и планировка зон наполнения должны предусматривать:
а) отдельные маркированные зоны для различных газов;
б) однозначное обозначение и разделение пустых баллонов (контейнеров) и баллонов (контейнеров), находящихся на разных стадиях производства (например, "Ожидает наполнения", "Наполнен", "Карантин", "Одобрен", "Отклонен").
Способ, используемый для достижения этих уровней разделения, зависит от характера, объема и сложности технологического процесса. Мерами разделения могут служить зоны с разметками пола, перегородки, барьеры, обозначения и т.д.
3.2 Оборудование
3.2.1 Все оборудование для производства и проведения анализов должно проходить периодическую аттестацию и калибровку (поверку).
3.2.2 Газ следует подавать только в предназначенный для него баллон (контейнер). Трубопроводы, по которым проходят газы, не должны иметь соединений, за исключением аттестованных процессов автоматического наполнения. Распределительные коллекторы должны быть оборудованы соединительными элементами для наполнения, соответствующими только клапану для конкретного газа или конкретной смеси газов так, чтобы к этому коллектору могли подсоединяться только соответствующие баллоны (контейнеры). Использование распределительных коллекторов и их соединений с клапанами баллонов (контейнеров) может регулироваться национальными или международными стандартами.
3.2.3 Работы по ремонту и техническому обслуживанию оборудования не должны влиять на качество медицинских газов.
3.2.4 Следует избегать наполнения немедицинских газов в зонах и на оборудовании, предназначенных для производства медицинских газов. Исключения допускаются в случаях, когда качество газа, используемого для немедицинских целей, по меньшей мере, эквивалентно качеству медицинского газа и требования настоящего стандарта соблюдаются. При этом для предотвращения загрязнения медицинского газа следует иметь аттестованный (валидированный) метод, который исключает проникание немедицинских газов в линию, обслуживающую зону наполнения.
3.2.5 Резервуары для хранения и передвижные цистерны для доставки должны быть предназначены для одного вида газа определенного качества. Сжиженные медицинские газы можно хранить или транспортировать в тех же резервуарах, что и аналогичные немедицинские газы, при условии, что качество последних, по меньшей мере, эквивалентно качеству медицинских газов.
4 Документация
4.1 Данные, включенные в протоколы для каждой серии наполненных баллонов (контейнеров), должны содержать информацию обо всех основных параметрах соответствующих стадий наполнения. Протоколы серии должны содержать следующую информацию:
- наименование продукции;
- дату и время операций по наполнению;
- ссылку на используемую установку для наполнения;
- используемое оборудование;
- название газа и ссылку на спецификацию для данного газа или на спецификации для каждого из газов, входящих в смесь;
- операции, предшествующие наполнению (7.3 данного приложения);
- количество и объем баллонов (контейнеров) до и после наполнения;
- фамилию и инициалы лица, проводившего операцию наполнения;
- фамилии и инициалы лиц, выполнявших каждую ответственную операцию (очистку линии, приемку баллонов (контейнеров), опорожнение баллонов (контейнеров) и т.п.);
- основные параметры, подтверждающие правильность наполнения при стандартных условиях;
- результаты испытаний при контроле качества и в случаях, когда оборудование калибруется перед каждым испытанием, спецификацию стандартного газа и результаты контроля калибровки;
- результаты контроля наполнения баллонов (контейнеров);
- образец этикетки с номером серии;
- подробную информацию о любых затруднениях или необычных случаях;
- утвержденное письменное разрешение на любое отклонение от инструкций по наполнению;
- дату и подпись контролера, ответственного за операцию наполнения, подтверждающую правильность проведения работы.
5 Производство
5.1 Все критические стадии технологических процессов подлежат аттестации (валидации).
5.2 Производство нерасфасованной продукции (балк-продукта)
5.2.1 Газы, предназначенные для медицинского применения, производят путем химического синтеза или получают из природных источников, при необходимости, с последующей очисткой (например, на установке разделения воздуха). Такие газы можно рассматривать как фармацевтические субстанции или нерасфасованные лекарственные средства.
5.2.2 При необходимости следует документально регламентировать требования к чистоте, другим составляющим и возможным примесям, которые могут присутствовать в исходном газе и на стадиях его очистки. Для каждого процесса должны быть разработаны соответствующие технологические схемы.
5.2.3 Все стадии разделения и очистки должны быть разработаны таким образом, чтобы обеспечить их максимальную эффективность. Например, примеси, которые могут оказать отрицательное влияние на стадию очистки, следует удалять до ее достижения.
5.2.4 Стадии разделения и очистки подлежат аттестации (валидации) в отношении эффективности и контролируются в соответствии с результатами аттестации (валидации). Внутрипроизводственный контроль может включать в себя непрерывное проведение анализов. Порядок обслуживания оборудования и замены расходных деталей (например, фильтров очистки) должен быть основан на результатах контроля и аттестации (валидации).
5.2.5 При необходимости в документации следует указывать предельные значения температуры в ходе технологических процессов, измеренной при внутрипроизводственном контроле.
5.2.6 Компьютерные системы, используемые для управления или непрерывного контроля процессов, должны быть аттестованы (валидированы).
5.2.7 Для непрерывного процесса следует документально определить понятие серии продукции, которое должно быть согласовано с порядком проведения анализа нерасфасованного газа.
5.2.8 В ходе процесса производства следует постоянно контролировать качество газа и содержание в нем примесей.
5.2.9 Следует проводить микробиологический контроль воды, используемой для охлаждения во время сжатия воздуха, если она имеет контакт с медицинским газом.
5.2.10 Все операции по транспортированию сжиженных газов из места первичного хранения, в т.ч. проведение контроля перед транспортированием, должны выполняться в соответствии с инструкциями, направленными на предотвращение любого загрязнения. Трубопровод для транспортирования газа должен быть оборудован обратным клапаном или другим аналогичным устройством. Особое внимание следует уделять очистке гибких соединительных элементов, шлангов и мест соединения.
5.2.11 Вновь поступивший газ можно добавлять в резервуары, в которых хранится этот же газ из предыдущих поставок. При этом результаты анализа пробы должны подтвердить, что качество поставленного газа соответствует установленным требованиям. Такая проба может быть отобрана из:
- поставленного газа перед добавлением или
- резервуара с нерасфасованным газом после добавления и смешения.
5.2.12 Нерасфасованные газы, предназначенные для медицинского применения, следует определять как серию продукции, контролировать согласно соответствующим требованиям и выдавать разрешение на их наполнение.
5.3 Наполнение и маркировка
5.3.1 До проведения операций наполнения следует определить показатели (параметры) серии продукции.
5.3.2 Баллоны (контейнеры) для медицинских газов должны соответствовать установленным техническим требованиям. После наполнения клапаны следует пломбировать для контроля первого вскрытия. Для обеспечения адекватной защиты от загрязнения рекомендуется устанавливать на баллоны клапаны удерживания остаточного давления.
5.3.3 Распределительный коллектор для наполнения медицинскими газами и баллоны (контейнеры) должны быть предназначены для одного определенного газа или смеси газов (3.2.2 данного приложения). Следует организовать систему контроля за оборотом баллонов и клапанов.
5.3.4 Очистку и продувку оборудования для наполнения и трубопроводов следует проводить в соответствии с утвержденными инструкциями. Эти операции имеют особое значение после технического обслуживания системы или нарушения ее целостности. Перед выдачей разрешения на использование систему необходимо проверить на отсутствие загрязнений. Следует вести и сохранять протоколы очистки оборудования.
5.3.5 Визуальное обследование баллонов (контейнеров) на производственном участке проводится в следующих случаях:
- при поступлении новых баллонов;
- до и после любого испытания гидростатическим давлением или эквивалентного испытания.
После установки клапана его следует держать в закрытом положении для предотвращения поступления любого загрязнения в баллон (контейнер).
5.3.6 Перед началом операции наполнения следует проводить:
- проверку остаточного давления (от 3 до 5 бар) для подтверждения того, что баллон (контейнер) полностью не опорожнен;
- изъятие баллонов (контейнеров), в которых остаточное давление не обнаружено, для проведения дополнительных мер, позволяющих установить, что эти баллоны (контейнеры) не содержат воду или другие загрязняющие вещества; эти действия могут включать в себя очистку с применением прошедших аттестацию (валидацию) методов или визуальный контроль;
- контроль поврежденных баллонов (контейнеров), с которых должны быть удалены этикетки, определяющие серию продукции, и все другие этикетки;
- визуальный контроль внешнего вида каждого клапана и баллона (контейнера) на наличие вмятин, прожогов от дуговой сварки, сколов, других повреждений и загрязнений маслами или смазками. Следует проводить надлежащим образом очистку, испытания и техническое обслуживание баллонов (контейнеров);
- проверку каждого баллона (контейнера) или клапанного соединения криогенного сосуда для определения соответствия его типа данному медицинскому газу;
- проверку "кода даты испытания" баллона (контейнера), удостоверяющую, что было проведено испытание баллона (контейнера) гидростатическим давлением (или эквивалентным испытанием) и срок действия результатов испытаний не истек в соответствии с действующими нормами;
- проверку наличия на каждом баллоне (контейнере) цветового кода по соответствующему стандарту.
5.3.7 Следует тщательно выполнять подготовку баллонов (контейнеров), возвращенных для повторного наполнения, чтобы риск загрязнения был минимальным. Для сжатых газов максимальное теоретическое содержание примеси при давлении наполнения 200 бар должно составлять 500 объемных частей на миллион. Для других давлений определяются эквивалентные значения.
Подготовка баллонов (контейнеров) выполняется следующим образом:
- из каждого баллона удаляется весь оставшийся газ путем откачивания (по крайней мере, до остаточного абсолютного давления 150 мбар) или
- в каждом баллоне (контейнере) сбрасывается давление и выполняется последующая продувка с использованием аттестованных (валидированных) методов (частичное создание избыточного давления не менее 7 бар с последующим сбросом).
Для баллонов (контейнеров), оборудованных клапанами остаточного (положительного) давления, достаточно одной откачки под вакуумом до давления 150 мбар. В противном случае следует проводить полный анализ оставшегося газа в каждом баллоне (контейнере).
5.3.8 Заполнение баллонов (контейнеров) следует проверять в установленном порядке. Одним из показателей того, что баллон (контейнер) наполняется надлежащим образом, может служить ощущение тепла при легком прикосновении к внешней оболочке баллона (контейнера) в ходе наполнения.
5.3.9 Каждый баллон должен иметь маркировку и цветовой код. Номер серии и/или дата наполнения, а также дата истечения срока годности могут быть указаны на отдельной этикетке.
6 Контроль качества
6.1 Качество воды, используемой для проведения испытаний гидростатическим давлением, должно соответствовать, по меньшей мере, качеству питьевой воды и подвергаться постоянному микробиологическому контролю.
6.2 Проведение испытаний и выдача разрешения на выпуск каждого медицинского газа должны осуществляться в соответствии с его спецификацией. Каждый медицинский газ должен испытываться на соответствие всем нормативным требованиям с интервалами, достаточными для гарантии постоянного соблюдения этих требований.
6.3 На проведение наполнения нерасфасованного газа должно выдаваться разрешение (5.2.12 данного приложения).
6.4 Если один медицинский газ наполняется через многоцилиндровый коллектор, то, по крайней мере, один баллон с газом от каждого наполнения через коллектор должен проверяться на подлинность, количественное содержание и, при необходимости, на содержание воды, при каждой смене баллонов (контейнеров) на коллекторе.
6.5 Если один медицинский газ наполняется в баллоны (контейнеры) путем индивидуальной операции наполнения, то, по крайней мере, один баллон (контейнер) при каждом непрерывном цикле наполнения должен быть проверен на подлинность и количественное содержание. Примером непрерывного цикла наполнения является производство в течение одной смены одним и тем же персоналом с использованием одного оборудования и одной серии газа в балк-форме.
6.6 Если медицинский газ готовится смешением в баллоне (контейнере) двух или более различных газов из одного и того же распределительного коллектора, то при каждом цикле работы коллектора следует испытывать содержимое, как минимум, одного баллона (контейнера) на подлинность и количественное содержание всех компонентов газовой смеси и, при необходимости, на содержание воды и правильность соотношения газов в смеси. Если баллоны (контейнеры) наполняются по отдельности, то каждый баллон (контейнер) следует испытывать на подлинность и количественное содержание всех компонентов газовой смеси, и, как минимум, один баллон (контейнер) из каждого непрерывного цикла наполнения - на правильность соотношения газов в смеси.
6.7 Если газы смешиваются на линии перед наполнением (например, смесь оксида азота/кислорода), то следует проводить постоянный анализ наполняемой смеси.
6.8 Если баллон (контейнер) наполняется несколькими газами, то процесс наполнения должен обеспечивать правильное смешивание газов в каждом баллоне (контейнере) и полную гомогенность смеси.
6.9 Каждый наполненный баллон (контейнер) перед опломбированием для контроля первого вскрытия должен проходить испытание на герметичность с использованием соответствующего метода. Следует также проверять на герметичность баллоны (контейнеры), из которых отбирались пробы для анализов.
6.10 Если криогенным газом наполняются криогенные сосуды для поставки потребителям, то содержимое каждого сосуда следует испытывать на подлинность и проводить его количественный анализ.
6.11 Не требуется отбирать пробы из криогенных сосудов после их наполнения в том случае, если они хранятся у потребителей и повторно заполняются на месте из передвижных цистерн, при условии, что компания, осуществляющая наполнение, предоставляет аналитический паспорт пробы, отобранной из такой цистерны. Криогенные сосуды, хранящиеся у потребителей, следует периодически испытывать для подтверждения соответствия их содержимого фармакопейным требованиям.
6.12 Не требуется сохранять образцы серий продукции, если это не предусмотрено документацией.
7 Хранение и выпуск
7.1 Наполненные баллоны (контейнеры) должны содержаться на карантинном хранении до выдачи разрешения на выпуск Уполномоченным лицом.
7.2 Баллоны (контейнеры) с газом следует хранить в специально отведенном месте, защищенном от воздействия экстремальных температур. Зоны хранения должны быть чистыми, сухими, хорошо проветриваемыми и в них не должны храниться горючие материалы, с целью сохранения баллонов (контейнеров) чистыми к моменту их использования.
7.3 Порядок хранения должен предусматривать раздельное хранение баллонов (контейнеров) с различными газами, наполненных и пустых баллонов (контейнеров), а также обеспечивать порядок оборота запасов согласно очередности поступления продукции на склад ("первым поступил - первым выдан").
7.4 Во время транспортирования газовые баллоны (контейнеры) должны быть защищены от неблагоприятных погодных условий. Для газовых смесей, в которых при замораживании происходит разделение фаз, должны соблюдаться особые условия хранения и транспортирования.
Термины и определения
баллон (cylinder): Транспортируемый контейнер вместимостью не более 150 л воды, находящийся под давлением (для целей данного приложения).
Примечание - В данном документе определение баллон подразумевает, в соответствующем контексте, понятие связка баллонов (упаковка баллонов).
газ (gas): Вещество или смесь веществ, которые при давлении 1,013 бар (101,325 кПа) и температуре 15 °С находятся полностью в газообразном состоянии или при 50 °С давление паров превышает 3 бар (300 кПа) (ISO 10286:1996 Gas cylinders - Terminology. Bilingual edition).
зона (area): Часть помещения, специально выделенная для производства медицинских газов.
испытание гидростатическим давлением (hydrostatic pressure test): Испытание, проводимое по соображениям безопасности в соответствии с требованиями национальных и международных норм для проверки того, что баллоны или резервуары могут выдержать высокие давления.
клапан удержания остаточного давления (minimum pressure retention valve): Клапан, снабженный обратной системой, поддерживающей определенное давление (около 3-5 бар выше атмосферного давления) и предотвращающей загрязнение клапана при использовании.
контейнер (container): Криогенный сосуд, цистерна, бак, баллон, связка баллонов или любая другая упаковка, которая вступает в непосредственный контакт с медицинским газом.
криогенный газ (cryogenic gas): Газ, который при давлении 1,013 бар сжижается при температуре ниже минус 150 °С.
криогенный сосуд (cryogenic vessel): Стационарный или переносной термически изолированный контейнер, сконструированный для хранения сжиженных или криогенных газов. Из криогенного сосуда газ отбирается в газообразном или жидком состоянии (для целей данного приложения).
максимальный теоретический остаточный уровень примеси (maximum theoretical residual impurity): Примесь газа от возможного предыдущего загрязнения, оставшаяся в баллонах перед наполнением после предварительной обработки.
Примечание - Вычисление максимального теоретического уровня примеси имеет значение только для сжатых газов в предположении, что эти газы ведут себя как идеальные.
медицинский газ (medicinal gas): Любой газ или смесь газов, предназначенные для введения больным в терапевтических, диагностических или профилактических целях для оказания фармакологического воздействия и классифицируемые как лекарственные средства.
нерасфасованный газ (bulk gas): Любой газ, предназначаемый для медицинского использования, который прошел все технологические стадии, за исключением стадии окончательной упаковки.
обратный клапан (non-return valve): Клапан, который позволяет проходить потоку только в одном направлении.
откачивать (evacuate): Удалять остаточный газ из контейнера с помощью вакуума.
продувка (purge): Операция по опорожнению и прочистке баллона:
- методом сброса и откачивания или
- методом сброса, частичного создания избыточного давления рассматриваемым газом и его последующим сбросом.
распределительный коллектор (manifold): Оборудование или устройство, сконструированные для одновременного опорожнения или наполнения одного или более контейнеров для газа.
резервуар (tank): Стационарный контейнер для хранения сжиженного или криогенного газа.
сброс (blowing down): Сброс давления до атмосферного.
связка баллонов (cylinder bundle): Сборка жестко закрепленных в каркасе баллонов, которые подсоединяются к распределительному коллектору, транспортируются и используются как один блок.
сжатый газ (compressed gas): Газ, который при наполнении под давлением остается полностью газообразным при температуре минус 50 °С (ISO 10286).
сжиженный газ (liquified gas): Газ, который при наполнении под давлением находится в двухфазном состоянии (газ над жидкостью) при температуре минус 50 °С.
установка разделения воздуха (air separation plant): Установка для отбора атмосферного воздуха и разделения его очисткой, фильтрацией, сжатием, охлаждением, сжижением и дистилляцией на газообразный кислород, азот и аргон.
цистерна (tanker): Передвижной контейнер для хранения сжиженного или криогенного газа.
Приложение 7
Производство лекарственных средств из растительного сырья
Общие положения
Лекарственные средства из растительного сырья имеют сложную природу, разнообразные характеристики и большое число активных ингредиентов, содержащихся в малых количествах. В связи с этим при производстве лекарственных средств из растительного сырья особую роль играет контроль исходных материалов, условий хранения и переработки.
Помещения
Складские зоны
1 Исходное растительное сырье (т.е. необработанные растения) следует хранить в отдельных помещениях. Эти помещения должны быть хорошо проветриваемыми и защищенными от проникания насекомых и животных, особенно грызунов. Следует предусмотреть меры против распространения животных и микроорганизмов, привносимых с растительным сырьем, а также против перекрестного загрязнения. Порядок размещения упаковок не должен препятствовать свободной циркуляции воздуха.
2 Особое внимание следует уделять обслуживанию и чистоте складских зон, в которых может образовываться пыль.
3 Для хранения растений, экстрактов, настоек и другой продукции могут потребоваться особые условия по влажности, температуре и освещенности, которые необходимо обеспечить и контролировать.
Производственные зоны
4 При отборе проб, взвешивании, смешении и других технологических операциях с растительным сырьем, сопровождающихся пылеобразованием, следует принимать особые меры по поддержанию чистоты, а также по предотвращению перекрестного загрязнения (удаление пыли, выделение специальных помещений и т.п.)
Документация
Спецификации на исходные материалы
5 Помимо данных, приведенных в настоящем стандарте (4.11), в спецификации на растительное сырье, используемое для производства лекарственных средств, следует, по возможности, включать:
- наименование, принятое в ботанике с указанием, при необходимости, классификатора (например, "Классификатор растений и животных" Карла Линнея);
- подробную информацию о происхождении растения (страна или местность, при необходимости, культура, время и способ сбора, использование пестицидов и др.);
- указание об использовании всего растения или только его части;
- данные о методе сушки, если приобретены высушенные растения;
- описание растения, а также данные его макро- и микроскопического исследований;
- данные об испытаниях на подлинность, в т.ч. испытания на подлинность для известных активных ингредиентов или маркеров; для определения подлинности необходимо иметь соответствующие образцы сравнения;
- по возможности, описание основных ингредиентов, обладающих установленной фармакологической активностью, или маркеров;
- методы определения содержания пестицидов и их допустимые концентрации;
- испытания на загрязнение грибами и/или бактериями (в т.ч. афлатоксины и пест-инфестацины) и пределы допустимого загрязнения;
- испытания на содержание тяжелых металлов и других возможных посторонних примесей;
- испытания на включение инородных материалов.
Любая обработка, направленная на уменьшение загрязнений грибами, бактериями и пр., должна быть документально оформлена. Такая документация должна включать в себя подробную информацию о процессе обработки, проводимых испытаниях и пределах остаточного загрязнения.
Технологические инструкции
6 Технологические инструкции должны содержать описание различных операций, проводимых с растительным сырьем (сушка, измельчение, просеивание и пр.), а также данные о времени и температуре сушки и методах, используемых для контроля размеров фрагментов или частиц. Они также должны содержать описание методов удаления посторонних материалов (например, просеивание и пр.).
Инструкции по производству лекарственных средств из растительного сырья должны включать в себя данные об их основе или растворителе, времени и температуре экстракции, подробное описание всех стадий концентрирования и используемых методов.
Отбор проб
7 Так как необработанное лекарственное сырье получают из отдельных растений, оно является неоднородным. Отбор проб следует проводить с особой предосторожностью специально обученным персоналом. Подлинность каждой серии должна подтверждаться отдельным документом.
Контроль качества
8 Персонал, занятый контролем качества, должен иметь специальную подготовку в области лекарственных средств из растительного сырья для того, чтобы он мог проводить испытания поставляемого растительного сырья на подлинность и наличие примесей, выявлять рост колоний грибов, заражение паразитами, неоднородность и т.п.
9 Подлинность и качество лекарственных средств из растительного сырья должны контролироваться в соответствии с нормативной документацией.
Приложение 8
Отбор проб исходных и упаковочных материалов
Общие положения
Отбор проб является важной операцией, при которой берется только малая часть материалов, предназначенных для производства всей серии. Нерепрезентативная проба не позволяет дать гарантированное заключение о качестве всей серии. Правильный отбор проб является одним из основных компонентов системы обеспечения качества.
Примечание - Порядок отбора проб приведен в пунктах 6.11-6.14 настоящего стандарта. В настоящем приложении приведены дополнительные указания по отбору проб исходных и упаковочных материалов.
Персонал
1 Персонал, занятый отбором проб, должен проходить начальное и систематическое повторное обучение по вопросам отбора проб. Программы обучения должны включать в себя:
- порядок отбора проб;
- письменные инструкции по отбору проб;
- методы и оборудование, используемые при отборе проб;
- методы предотвращения перекрестного загрязнения;
- меры предосторожности при работе с нестабильными и/или стерильными веществами;
- важность учета внешнего вида материалов, упаковок и этикеток;
- необходимость протоколирования любых непредвиденных или необычных обстоятельств.
Исходные материалы
2 Подлинность всей серии исходных материалов может быть гарантирована только при взятии проб из всех упаковок и проведении теста на подлинность каждой пробы. Допускается отбор проб из части упаковок при наличии аттестованной методики, исключающей возможность неправильной маркировки каждой упаковки с исходными материалами.
3 При аттестации такой методики следует учитывать:
- данные о производителе и поставщике, а также уровень выполнения ими требований настоящего стандарта;
- систему обеспечения качества предприятия - производителя исходных материалов;
- условия производства и контроля исходных материалов;
- характер и свойства исходных материалов, а также получаемой из них продукции.
По этим данным можно аттестовать методику, допускающую отбор проб не из каждой упаковки с исходными материалами при выполнении следующих условий:
- исходные материалы поступают от одного производителя или с одного предприятия;
- исходные материалы поступают непосредственно от производителя или в опечатанной производителем упаковке, причем этот поставщик имеет безупречную репутацию, а аудит его системы обеспечения качества регулярно проводится покупателем (производителем лекарственных средств) или официально Уполномоченным лицом.
Такую методику нельзя аттестовать в следующих случаях:
- исходные материалы поступают от посредников и производитель неизвестен или не прошел аудит;
- исходные материалы используются для производства парентеральной продукции.
4 Качество серии исходных материалов может быть оценено путем отбора репрезентативной пробы и проведения испытаний. Для этой цели могут использоваться пробы, взятые для проведения теста на подлинность. Количество образцов, отбираемых для получения репрезентативной пробы, определяется статистическим методом и указывается в плане отбора проб. Количество отдельных образцов, которые могут быть смешаны для получения общей пробы, определяется с учетом вида материала, данных о поставщике, а также его однородности.
Упаковочные материалы
5 При составлении плана отбора проб упаковочных материалов необходимо принимать во внимание их количество, требуемое качество, характер материалов (например, первичные упаковочные материалы и/или печатные упаковочные материалы), методы производства, а также информацию о системе обеспечения качества у производителя упаковочных материалов, полученную при проведении аудита. Количество отбираемых проб определяется статистически и указывается в плане отбора проб.
Приложение 9
Производство жидкостей, кремов и мазей
Общие положения
В процессе производства жидкостей, кремов и мазей следует принимать особые меры предосторожности ввиду их предрасположенности к микробному и иному загрязнению. Поэтому необходимо принимать специальные меры по предупреждению любого вида загрязнения.
Помещения и оборудование
1 Для защиты от загрязнения при производстве и перемещении продукции рекомендуется использовать закрытые системы. Производственные зоны, в которых находится открытая продукция или открытые чистые упаковки, как правило, следует оборудовать эффективной системой вентиляции, имеющей фильтры очистки воздуха.
2 Конструкция и расположение реакторов, емкостей, трубопроводов и насосов должны предусматривать удобство их очистки и, при необходимости, дезинфекции. В частности, в конструкции оборудования должно быть сведено к минимуму наличие мертвых зон и тупиков, в которых могут концентрироваться остатки материалов, вызывающие размножение микроорганизмов.
3 По возможности, не рекомендуется использовать оборудование из стекла. Во многих случаях детали оборудования, входящие в контакт с продукцией, должны изготавливаться из высококачественной нержавеющей стали.
Производство
4 Следует задавать требования к химическому и микробиологическому качеству воды, используемой в производстве, и обеспечивать контроль выполнения этих требований. Во избежание риска размножения микроорганизмов следует надлежащим образом организовать обслуживание систем подготовки воды. После обработки систем подготовки воды химическими средствами их необходимо промыть по аттестованной (валидированной) методике, гарантирующей полное удаление дезинфицирующих веществ.
5 Качество материалов, получаемых в цистернах, следует проверять до их перемещения в емкости для хранения.
6 Следует контролировать передачу материалов по трубопроводам, чтобы гарантировать их поступление в нужное место.
7 В помещениях, где содержится открытая продукция или чистые упаковки, не допускается нахождение материалов, способствующих выделению волокон и других загрязняющих веществ (например, из картонных или деревянных поддонов).
8 Во время операций наполнения следует обеспечивать однородность (гомогенность) смесей, суспензий и т.п. Процесс перемешивания и наполнения подлежит аттестации (валидации). Особое внимание необходимо уделять обеспечению однородности смеси в начале, после остановок и в конце процесса наполнения.
9 Если готовый продукт упаковывается не сразу после окончания производственных операций, необходимо установить максимально допустимое время до его упаковки и соответствующие условия хранения.
Приложение 10
Производство аэрозолей для ингаляций
Производство аэрозолей для ингаляций, наполняемых в баллоны, снабженных дозирующими клапанами и находящихся под давлением, обладает характерными особенностями и к нему предъявляются специальные требования. Условия производства должны обеспечивать максимальную защиту продукта от загрязнения микроорганизмами и частицами. Особое значение имеет качество деталей клапанов, а в случае суспензий - их однородность.
Общие положения
1 Как правило, используются два метода производства и наполнения:
a) двухступенчатый метод (наполнение под давлением).
Активный ингредиент вводится в пропеллент с высокой точкой температуры кипения, упаковка наполняется дозой активного ингредиента, на нее надевается клапан и пропеллент с низкой точкой температуры кипения вводится через отверстие клапана. Температура активного ингредиента поддерживается низкой во избежание потерь за счет испарения;
b) одноступенчатый метод (холодное наполнение).
Активный ингредиент вводится в смесь пропеллентов под высоким давлением и/или при низкой температуре. Затем производится наполнение упаковки в один прием.
Помещения и оборудование
2 Производство и наполнение рекомендуется проводить, по возможности, в закрытых системах.
3 Там, где продукция или ее чистые компоненты содержатся открытыми, помещения должны соответствовать, как минимум, зоне D. Воздух в них должен подаваться через фильтры, а доступ осуществляться через воздушные шлюзы.
Производство и контроль качества
4 Дозирующие клапаны для аэрозолей являются более сложными, чем большинство устройств, используемых в фармацевтической промышленности. Технические условия на них, методики отбора образцов и контроль дозирующих клапанов должны соответствовать требованиям, предъявляемым к качеству такой продукции. Особое значение имеет проведение аудита системы обеспечения качества у производителя дозирующих клапанов.
5 Все летучие вещества (например, жидкие или газообразные пропелленты) должны быть пропущены через фильтр, удерживающий частицы с размерами более 0,2 мкм. Рекомендуется, по возможности, проводить дополнительную фильтрацию непосредственно перед наполнением.
6 Упаковки и клапаны должны проходить очистку в соответствии с аттестованной (валидированной) методикой, учитывающей свойства продукции и обеспечивающей отсутствие загрязнения вспомогательными технологическими средствами (например, смазочными), а также микробного загрязнения. После очистки клапаны следует хранить в чистых закрытых упаковках. Во избежание внесения загрязнения при последующих операциях (например, при отборе проб для проведения испытаний) необходимо принимать меры предосторожности. Упаковки должны поступать на линию наполнения очищенными или очищаться на линии непосредственно перед наполнением.
7 Необходимо обеспечить однородность суспензии в точке наполнения в ходе всего процесса наполнения.
8 При использовании двухступенчатого метода для получения заданной смеси необходимо обеспечить точную массу вводимых веществ на обоих этапах. Поэтому во многих случаях целесообразен 100%-ный контроль массы на каждом этапе.
9 Контроль после наполнения должен подтвердить отсутствие утечек. Все проверки на наличие утечек следует проводить так, чтобы не допускать микробного загрязнения или остаточной влаги.
Приложение 11
Системы с компьютерным управлением и контролем
Основные положения
Внедрение систем с компьютерным управлением и контролем в производстве лекарственных средств, в т.ч хранение, распределение и контроль качества, не должно приводить к нарушению принципов настоящего стандарта. Замена ручных операций системами с компьютерным управлением и контролем не должна приводить к ухудшению качества продукции или нарушениям в системе обеспечения качества. Необходимо учесть риск потери отдельных свойств предыдущего порядка работы, связанный с уменьшением роли операторов.
Персонал
1 Важную роль играет тесное взаимодействие руководящих работников и сотрудников, занятых с системами с компьютерным управлением и контролем. Лица, занимающие ответственные должности, должны пройти необходимую подготовку для работы в рамках их полномочий с системами, в которых используются компьютеры. Это должно обеспечить достаточно высокий уровень экспертизы при проектировании, монтаже, аттестации (валидации) и эксплуатации систем с компьютерным управлением и контролем.
Аттестация
2 Объем необходимой аттестации зависит от назначения применяемой системы, вида аттестации (перспективной или ретроспективной) и введения в систему новых элементов. Аттестация должна рассматриваться как неотъемлемая часть всего цикла жизни системы, включающего в себя этапы разработки, программирования, испытаний, сдачи в эксплуатацию, документального оформления, функционирования, контроля и модернизации.
Требования к системе
3 Оборудование следует размещать в надлежащих условиях, исключающих влияние посторонних факторов на работу системы.
4 Следует разработать и регулярно обновлять детальное описание системы (в т.ч. соответствующих схем). Это описание должно включать в себя общие положения, назначение, состав системы, меры безопасности, область применения, характеристики использования компьютера и его взаимодействие с другими системами и процессами.
5 Программное обеспечение является основой компьютерных систем. Пользователь должен убедиться в соответствии программного обеспечения требованиям системы качества.
6 Система с компьютерным управлением и контролем должна включать в себя, по возможности, встроенные программы проверки правильности ввода и обработки данных.
7 Перед началом работы необходимо провести тщательную проверку системы с компьютерным управлением и контролем и убедиться в ее соответствии заданным требованиям. При замене ручной системы на систему с компьютерным управлением и контролем следует обеспечить их параллельную работу в течение некоторого времени, рассматривая это как часть процедуры контроля и аттестации.
8 Ввод данных и их корректировка могут выполняться только сотрудниками, имеющими для этого доступ. Для предотвращения несанкционированного ввода данных следует использовать специальные ключи, идентификационные карты, персональные коды и др. ограничители доступа к компьютерным терминалам. Должна существовать процедура выдачи, отмены и изменения права доступа к вводу и исправлению данных, в т.ч. периодическая смена личных паролей. По возможности следует применять системы, позволяющие регистрировать несанкционированные попытки входа в систему.
9 При ручном вводе критических данных (например, массы или номера серии ингредиента при взвешивании) необходимо дополнительно проверить правильность ввода данных. Эта проверка может быть проведена другим оператором или аттестованными электронными средствами.
10 Система должна регистрировать имена операторов, вводящих или подтверждающих ввод критических данных. Допуск к исправлению введенных данных должен быть ограничен узким кругом лиц. Внесение любого изменения, касающегося ввода данных, требует специального допуска и должно протоколироваться с указанием причины изменений. Необходимо рассмотреть возможность включения в систему формирования протокола всех операций по вводу и исправлению данных.
11 Изменения в системе или в программном обеспечении должны проводиться в соответствии с определенной процедурой, включающей в себя действия по аттестации, проверке, согласованию и окончательному внесению изменений. Изменения должны вноситься только по согласованию с лицом, ответственным за конкретный участок системы, и оформляться документально. Все существенные изменения подлежат аттестации.
12 Для проведения аудита качества необходима возможность получения четких распечатанных копий данных, хранящихся в электронном виде.
13 Данные должны быть защищены физическими или электронными методами от преднамеренного или случайного уничтожения в соответствии с 4.9 настоящего стандарта. Необходимо проверять надежность и точность данных и порядок доступа к ним. Если планируется внесение изменений в компьютерное оборудование или программное обеспечение, упомянутые проверки должны проводиться с периодичностью, определяемой с учетом применяемых носителей информации.
14 Данные должны быть защищены путем их регулярного копирования. Копии следует хранить в течение установленного периода времени в изолированном и безопасном помещении.
15 На случай отказов в системе с компьютерным управлением и контролем должно быть предусмотрено необходимое резервное оборудование. Время приведения резервного оборудования в действие определяется допустимым интервалом в работе системы. Например, информация, необходимая для отзыва продукции, должна быть получена как можно быстрее.
16 Следует разработать и аттестовать порядок действия на случай остановки или неисправности системы. Все неисправности и меры по их устранению должны быть оформлены документально.
17 Порядок ведения документации, анализа ошибок и корректирующих действий должен быть определен специальной инструкцией.
18 При обслуживании компьютерного оборудования другой организацией необходимо заключить с ней официальный договор, в котором следует четко определить ответственность этой организации (раздел 7 настоящего стандарта).
19 В случаях, когда выпуск серии продукции осуществляется с использованием системы с компьютерным управлением и контролем, эта система должна предусматривать допуск к выпуску серии продукции только Уполномоченного лица и обеспечивать при этом его четкую идентификацию.
Приложение 12
Использование ионизирующего излучения в производстве
лекарственных средств
Примечание - Производитель продукции, для которой ионизирующее облучение является составной частью технологического процесса, должен также руководствоваться нормативными документами, регламентирующими использование ионизирующего излучения при производстве лекарственных средств.
Введение
Ионизирующее излучение может использоваться в производственном процессе для снижения степени биозагрязнения, стерилизации исходных материалов, упаковки или изделий, обработки препаратов на основе крови и других целей.
Существует два вида радиационных установок: с радионуклидными источниками гамма-излучения и с ускорителями электронов высокой энергии.
Гамма-установки
Возможны два режима обработки:
а) порционный: продукция размещается в фиксированном положении вокруг источника излучения и не может быть загружена или выгружена во время облучения;
б) непрерывный: автоматизированная транспортная система доставляет продукцию в камеру для облучения, перемещает ее с заданной скоростью по заданному маршруту, а затем выводит из камеры.
Радиационные установки с ускорителями электронов
Продукцию перемещают под непрерывным или импульсным пучком электронов высокой энергии, который сканируют в обоих направлениях перпендикулярно к перемещению продукции.
Ответственность
1 Радиационную обработку продукции может проводить непосредственно ее производитель или по контракту фирма, имеющая в распоряжении радиационную установку. При этом они должны иметь разрешения, оформленные в установленном порядке.
2 Производитель фармацевтической продукции несет ответственность за качество продукции, в т.ч. за результаты воздействия ионизирующего излучения. Фирма, проводящая радиационную обработку, несет ответственность за то, чтобы каждая упаковка получила дозу, определенную производителем (в т.ч. упаковка с продукцией, самая удаленная от источника излучения).
3 Требуемая поглощенная доза с указанием установленных пределов должна быть указана в технической документации на продукцию.
Дозиметрия
4 Дозиметрия - измерение поглощенной дозы ионизирующего излучения с помощью дозиметров. Понимание принципов работы и правильное использование техники важны для аттестации (валидации), наладки и контроля процесса.
5 Калибровка каждой партии рабочих дозиметров должна быть прослеживаемой вплоть до национального или международного эталонов. Установленную периодичность калибровки следует строго соблюдать.
6 Для определения изменения поглощения штатных дозиметров после облучения как при калибровке, так и при проведении дозиметрии должен использоваться один и тот же прибор. При использовании разных приборов они должны быть калиброваны в абсолютных единицах поглощения.
7 В зависимости от типа используемых дозиметров необходимо учитывать возможные источники погрешности, зависящие от влажности, температуры, периода времени между облучением и измерением, а также мощности поглощенной дозы.
8 Приборы, используемые для измерения изменения поглощения дозиметров по длине волны и для измерения толщины дозиметров, необходимо калибровать. Периодичность калибровки зависит от назначения, стабильности и способа применения.
Аттестация (валидация) процесса
9 Аттестация (валидация) - действие, доказывающее, что процесс, т.е. получение продукцией заданной поглощенной дозы, достигает ожидаемых результатов. Требования к аттестации (валидации) приведены в ГОСТ Р ИСО 11137-2000 "Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукции" и других нормативных документах.
10 Аттестация (валидация) должна включать в себя составление карты дозного поля, чтобы установить распределение поглощенной дозы внутри облучаемого контейнера с продукцией при определенной конфигурации упаковки продукции.
11 Спецификации на процесс облучения должны включать в себя:
a) описание упаковки продукции;
b) конфигурацию(и) укладки продукции внутри контейнера. Если в контейнере находится продукция различных видов, необходимо обратить особое внимание на возможное недооблучение продукции с высокой плотностью или затенение других изделий такой продукцией. Каждый способ укладки в контейнер разных видов продукции должен быть описан в технической документации и аттестован (валидирован);
c) конфигурацию расположения контейнеров вокруг источника (порционный режим) или маршрут облучаемых объектов внутри камеры для облучения (непрерывный режим);
d) верхний и нижний пределы допускаемых значений поглощенной дозы в продукции (и соответствующие показания дозиметров);
e) верхний и нижний пределы значений поглощенной дозы в продукции по всему объему контейнера и соответствующие методы дозиметрии для контроля этих значений;
f) другие параметры процесса, в т.ч. мощность поглощенной дозы, максимальное время облучения, количество циклов облучения и т.д.
Если облучение проводится по контракту третьей стороной, в контракт должны быть включены, как минимум, перечисления d) и е).
Аттестация установки
Общие положения
12 Аттестация заключается в экспериментальном получении и документировании фактов, доказывающих, что радиационная установка способна в течение длительного времени функционировать в установленных пределах согласно документации на процесс. Согласно данному приложению, установленные пределы - максимальное и минимальное допускаемые значения поглощенной дозы в продукции (по всему объему облучаемого контейнера). Изменения в работе установки, которые могут привести к выходу значений поглощенной дозы за эти пределы, ни при каких условиях не должны происходить без ведома оператора.
13 Аттестация должна включать в себя:
a) основные параметры установки;
b) карту дозного поля;
c) документирование;
d) требования к повторному вводу в эксплуатацию.
Гамма-установки
14 Основные параметры
Поглощенная доза в продукции зависит от следующих основных факторов:
a) активности и геометрии источника излучения;
b) расстояния от источника до контейнера;
c) продолжительности облучения, контролируемой таймером, или скоростью движения конвейера;
d) состава и плотности материала, а также наличия другой продукции между источником и рассматриваемой частью контейнера.
15 Суммарная поглощенная доза зависит также от маршрута, по которому движутся контейнеры при непрерывном режиме облучения, или от конфигурации загрузки при порционном режиме облучения и количества циклов облучения.
16 При фиксированном маршруте (при непрерывном облучении) или при фиксированной конфигурации загрузки (при порционном режиме облучения), а также для постоянной мощности источника и вида продукции основным параметром установки, контролируемым оператором, является скорость конвейера или время, установленное на таймере.
Карта дозного поля
17 При определении карты дозного поля камера для облучения должна быть заполнена контейнерами с муляжами продукции или образцами продукции однородной плотности. Дозиметры должны быть расположены не менее чем в трех заполненных контейнерах, окруженных аналогичными контейнерами или муляжами продукции. Если продукция уложена неравномерно, дозиметры должны быть размещены в большем количестве контейнеров.
18 Расположение дозиметров зависит от размеров облучаемого контейнера. Например, для контейнеров размером 1,0х1,0х0,5 м подходит расположение дозиметров в узлах трехмерной решетки с шагом 20 см, с учетом внешней поверхности контейнера. Если предполагаемые зоны с максимальной и минимальной дозами известны из предыдущих опытов, то часть дозиметров может быть изъята из зон со средними значениями доз и помещена в зоны с экстремальными значениями дозы с шагом 10 см.
19 В результате этой процедуры должны быть определены минимальная и максимальная поглощенные дозы, полученные продукцией в контейнере и на его поверхности для заданной комбинации параметров установки и облучения, плотности и конфигурации укладки.
20 В идеальном случае для определения карты дозного поля следует использовать эталонные дозиметры, поскольку они имеют наибольшую точность. Допускается использовать штатные дозиметры, при этом в зонах с предполагаемыми максимальными и минимальными значениями поглощенной дозы облучения, а также в точках, предназначенных для контроля процесса, возле штатных дозиметров рекомендуется помещать эталонные дозиметры. Полученные значения поглощенной дозы будут иметь случайную погрешность, которая может быть определена при многократных измерениях.
21 Минимальная наблюдаемая доза, измеряемая штатными дозиметрами, необходимая для получения всеми контейнерами минимальной требуемой дозы, должна устанавливаться с учетом случайной погрешности используемых штатных дозиметров.
22 Во время определения карты дозного поля параметры установки необходимо поддерживать постоянными, контролировать и регистрировать. Эти записи вместе с результатами дозиметрии и другими полученными протоколами следует сохранять.
Радиационные установки с ускорителями электронов
23 Основные параметры установки
Поглощенная доза ионизирующего излучения в продукции зависит от следующих основных факторов:
a) характеристики пучка (энергии электронов, среднего тока пучка, ширины развертки и неравномерности пучка по ширине развертки);
b) скорости конвейера;
c) состава и плотности продукции;
d) состава, плотности и толщины материала, находящегося между выходным окном и облучаемой частью продукции;
е) расстояния от выходного окна до контейнера.
24 Основными параметрами, контролируемыми оператором, являются характеристики пучка и скорость конвейера.
25 Карта дозного поля
При определении карты дозного поля дозиметры следует располагать между слоями гомогенного поглотителя, имитирующего реальную продукцию, или между слоями реальной продукции однородной плотности так, чтобы не менее 10 измерений были проведены в пределах максимального пробега электронов (пункты 18-21 данного приложения).
26 При определении карты дозного поля по объему облучаемого объекта параметры радиационной установки необходимо поддерживать постоянными, контролировать и регистрировать. Следует сохранять эти записи вместе с результатами дозиметрии и другими данными.
Процедура повторного ввода установки в эксплуатацию
27 Ввод в эксплуатацию должен проводиться заново всякий раз, когда происходят изменения процесса или параметров радиационной установки, способные привести к изменению распределения поглощенной дозы в контейнере для облучения (например, при замене облучателя радиационной установки). Объем работ по повторному вводу в эксплуатацию зависит от степени изменений, внесенных в конструкцию облучателя радиационной установки или конфигурацию загрузки. При наличии сомнений процедуру повторного ввода установки в эксплуатацию следует провести заново.
Помещения
28 Во избежание перекрестного загрязнения в помещении должны быть предусмотрены меры по разделению облученных и необлученных контейнеров. Если материалы находятся в закрытых контейнерах и отсутствует риск перекрестного загрязнения, нет необходимости при облучении отделять фармацевтические материалы от нефармацевтических. Любая возможность загрязнения продукции радионуклидами должна быть исключена.
Технологический процесс
29 Контейнеры с продукцией должны устанавливаться в соответствии с конфигурацией загрузки, определенной в процессе аттестации (валидации).
30 В ходе облучения поглощенная доза на поверхности контейнера должна контролироваться в соответствии с аттестованными (валидированными) дозиметрическими методиками. Зависимость между этой дозой и дозой, поглощенной продукцией внутри контейнера, должна быть установлена при аттестации (валидации) радиационной установки.
31 Для того чтобы различать облученные и необлученные контейнеры, можно использовать индикаторы ионизирующего излучения. Однако они не должны использоваться в качестве единственного средства, определяющего правильность проведения процесса.
32 Одновременную обработку разных видов продукции в одной загрузке следует проводить только тогда, когда по результатам эксплуатации установки или по другим данным известно, что поглощенная доза в каждом виде продукции находится в установленных пределах.
33 Получение требуемой поглощенной дозы излучения за несколько циклов облучения должно быть согласовано между производителем и организацией, проводящей облучение. Полная доза облучения должна набираться в течение предварительно установленного интервала времени. Производитель должен быть уведомлен о фактах незапланированных перерывов между циклами облучения, если продолжительность перерывов превышает ранее согласованные значения.
34 Облученная продукция всегда должна быть отделена от необлученной. Способы достижения этого включают в себя использование индикаторов радиации (пункт 31) и соответствующей планировки помещений (пункт 28).
Гамма-излучение
35 При непрерывном режиме облучения дозиметры должны быть расположены таким образом, чтобы под воздействием излучения одновременно находились не менее двух дозиметров.
36 При порционном режиме в точке контроля минимального значения поглощенной дозы облучения должны быть расположены не менее двух дозиметров.
37 При непрерывном режиме облучения должна быть предусмотрена индикация требуемого рабочего положения источника. Положение источника и движение конвейера должны быть связаны блокировкой. Скорость движения конвейера необходимо постоянно контролировать и регистрировать.
38 При порционном режиме облучения перемещение источника и продолжительность каждого облучения продукции должны контролироваться и регистрироваться.
39 Следует корректировать время облучения и скорости движения конвейера с учетом распада или дозарядки источника излучения. Периодичность аттестации (валидации) параметров установки или скорости конвейера должна соответствовать параметрам излучателя.
Радиационная установка с ускорителем электронов
40 Дозиметр должен быть размещен на каждом контейнере в контрольной точке.
41 Необходимо непрерывно регистрировать среднее значение тока пучка, энергию электронов, ширину развертки и скорость конвейера. Эти параметры, за исключением скорости конвейера, должны проверяться через определенные интервалы времени, установленные при аттестации, поскольку они подвержены спонтанному изменению.
Документация
42 Количество поступивших контейнеров и контейнеров, прошедших облучение и вывезенных с предприятия, должно совпадать и соответствовать значениям, указанным в сопроводительной документации. Любые расхождения должны протоколироваться и расследоваться.
43 Оператор радиационной установки должен подтверждать в письменном виде диапазон значений поглощенных доз, полученных каждым контейнером при каждой загрузке или в серии продукции.
44 Технологические протоколы и протоколы контроля для каждой серии продукции, прошедшей облучение, должны проверяться и подписываться специально назначенным лицом и сохраняться на предприятии. Метод и место хранения определяются по договоренности между фирмой, проводившей облучение, и производителем.
45 Документация, относящаяся к аттестации (валидации) радиационной установки, должна сохраняться в течение одного года после окончания срока годности продукции или в течение пяти лет после выпуска на реализацию последней продукции, прошедшей облучение на этой установке, в зависимости от того, какой период дольше.
Микробиологический контроль
46 Ответственность за проведение микробиологического контроля, в т.ч. за контроль окружающей среды в месте производства продукции и контроль продукции перед облучением, проводимый в соответствии с нормативной документацией, возлагается на производителя лекарственных средств.
Приложение 13
Производство лекарственных средств для клинических исследований
Введение
Лекарственные средства, предназначенные для проведения клинических исследований, производятся, как правило, в лабораторных условиях по нестандартным методикам. Настоящее приложение является руководством по составлению заказов, доставке и возврату лекарственных средств для клинических исследований.
Термины и определения
слепой метод (blinding): Процедура, при которой одна или более сторон, участвующих в клиническом исследовании, держатся в неведении относительно цели(ей) данного исследования. Единичный слепой метод означает неосведомленность субъектов исследования, а двойной слепой метод - неосведомленность о целях клинического исследования субъектов исследования, исследователей, наблюдателей и, в некоторых случаях, лиц, анализирующих полученные данные.
клиническое исследование (clinical trial): Исследование, проводимое с участием человека в качестве субъекта для выявления или подтверждения клинических, фармакологических и/или других фармакодинамических эффектов исследуемого препарата(ов), и/или нежелательных реакций на исследуемый(е) лекарственный(е) препарат(ы), и/или изучения его(их) всасывания, распределения, метаболизма и выведения с целью оценки его(их) безопасности и/или эффективности.
препарат сравнения (comparator product): Разрабатываемое или имеющееся на рынке лекарственное средство (положительный контроль) или плацебо, используемое в качестве препарата сравнения при проведении клинических исследований.
лекарственное средство для клинических исследований (investigational product): Дозированная лекарственная форма активной фармацевтической субстанции или плацебо, исследуемая или используемая в качестве препарата сравнения при проведении клинического исследования, включая уже зарегистрированные лекарственные препараты, если способ их применения или производства (лекарственная форма или упаковка) отличается от зарегистрированной формы, или в случае их использования по еще не одобренным показаниям, или для получения дополнительной информации об уже зарегистрированной дозированной лекарственной форме.
исследователь (investigator): Лицо, ответственное за проведение клинического исследования в месте его выполнения. При проведении клинического исследования группой лиц исследователем является руководитель группы.
заказ (order): Задание на приготовление, упаковку и/или доставку определенного количества единиц лекарственного средства для клинических исследований.
досье на лекарственное средство (product specification file): Комплект документов, содержащих всю информацию, необходимую для составления подробных инструкций по изготовлению, упаковке, контролю качества, выдаче разрешения на реализацию серии и доставке лекарственного средства.
отгрузка (shipping/dispatch): Операции по упаковке и отправке заказанных лекарственных средств для клинических исследований.
спонсор (sponsor): Физическое или юридическое лицо, являющееся инициатором клинического исследования и несущее ответственность за его организацию и финансирование.
Управление качеством
1 К аттестации (валидации) некоторых технологических процессов, применяемых при производстве лекарственных средств для клинических исследований, не имеющих регистрации, не могут быть предъявлены такие же требования, как при производстве зарегистрированных лекарственных средств. Для стерильных препаратов аттестация (валидация) процессов стерилизации должна проводиться в том же объеме, что и для зарегистрированных лекарственных препаратов. Спецификации и технологические инструкции на лекарственные средства для клинических исследований могут изменяться в процессе их разработки, что предъявляет особые требования к наличию высокоэффективной системы обеспечения качества.
2 Система обеспечения качества, разработанная производителем, должна учитывать требования настоящего стандарта, относящиеся к лекарственным средствам для клинических исследований, и должна быть документально оформлена и утверждена спонсором клинического исследования.
3 Операции по упаковке и маркировке лекарственных средств для клинических исследований часто выполняются после выдачи разрешения на реализацию серии нерасфасованной продукции в соответствии с конкретными требованиями исследований и имеют первостепенное значение для целостности клинических исследований. В связи с этим в соответствии с нормативным документом, регламентирующим правила проведения клинических исследований, и пунктом 9.2 настоящего стандарта проведение самоинспекции или независимого аудита является неотъемлемой составной частью системы обеспечения качества.
Персонал
4 Несмотря на небольшое количество персонала, обычно занятого в производстве лекарственных средств для клинических исследований, должны быть назначены отдельные сотрудники, ответственные за производство и контроль качества. Все производственные операции должны выполняться под руководством четко определенного ответственного лица. Персонал, дающий разрешение на выпуск лекарственных средств для клинических исследований, должен обладать необходимыми знаниями по вопросам системы контроля качества, требований настоящего стандарта и нормативных документов, относящихся к данному виду продукции. Лица, ответственные за выпуск продукции, должны быть независимыми от лиц, отвечающих за производство.
Помещения и оборудование
5 При производстве лекарственных средств для клинических исследований в одном помещении одновременно могут проводиться операции с разными продуктами. В связи с этим повышаются требования к защите от риска загрязнения, в т.ч. от перекрестного загрязнения и перепутывания продуктов, путем использования соответствующих инструкций.
6 Производство отдельных видов продуктов, перечисленных в пункте 3.6 настоящего стандарта, может быть организовано не в специальных помещениях и оборудовании, а по принципу отдельных циклов производства. Поскольку токсичность веществ не всегда известна, особое внимание следует уделять методикам очистки. При этом следует учитывать растворимость препаратов и вспомогательных веществ в различных растворах, предназначенных для очистки.
7 Аттестация (валидация) стерильных процессов при небольшом объеме серии связана с некоторыми трудностями. В этих случаях количество наполненных единиц может совпадать с максимальным количеством единиц, наполняемых при производстве. Наполнение и герметизация могут выполняться вручную, что представляет опасность для стерильности продукции. В этом случае особое внимание следует уделять контролю состояния окружающей среды.
Документация
8 Спецификации (на исходные материалы, первичные упаковочные материалы, промежуточные продукты, нерасфасованную и готовую продукцию), промышленные регламенты, технологические инструкции, а также инструкции по упаковке продукции могут изменяться по мере разработки препарата. Каждая новая версия документа должна учитывать последние данные, используемую технологию, нормативные и фармакопейные требования со ссылкой на предыдущую версию для обеспечения прослеживаемости. Причины внесения изменений следует протоколировать.
9 В случаях, когда отсутствует необходимость в составлении промышленного регламента и технологических инструкций, следует для каждой производственной операции или поставки составлять инструкции и протоколы, оформленные в письменном виде. Протоколы имеют особенно важное значение при переходе к серийному производству и подготовке окончательного комплекта документации.
10 Протоколы производства серий лекарственных средств следует хранить не менее двух лет после завершения или официального прекращения клинических исследований или в течение периода времени, установленного в соответствующей документации.
Заказ
11 Заказ может включать в себя производство и/или упаковку некоторого числа единиц продукции и/или их отгрузку. Производителю лекарственных средств для клинических исследований заказ может выдавать только спонсор клинического исследования. Заказ должен составляться в письменном виде (но может передаваться и электронным способом) и быть достаточно четким во избежание разночтений. Заказ должен быть официально утвержден и иметь ссылку на утвержденное досье на лекарственное средство.
Досье на лекарственное средство
12 Вся информация, необходимая для составления подробных письменных инструкций по производству, упаковке, контролю качества, выпуску серии продукции, хранению и/или отгрузке, должна содержаться в досье на лекарственное средство. Досье на лекарственное средство должно непрерывно обновляться, обеспечивая логическую связь с предыдущими версиями.
Промышленные регламенты и технологические инструкции
13 Любые изменения в промышленный регламент и технологические инструкции следует вносить в соответствии с инструкцией, направленной на оценку влияния этого изменения на стабильность и биоэквивалентность лекарственного средства. Все изменения должны быть оформлены протоколом и утверждены ответственными лицами.
Инструкции по упаковке
14 Упаковка и маркировка лекарственных средств для клинических исследований, как правило, являются более сложными операциями в связи с большей опасностью возникновения ошибок (которые труднее выявить), чем упаковка зарегистрированных лекарственных средств, так как при этом используются "слепые" этикетки. В связи с этим необходимо уделять особое внимание учету этикеток, контролю чистоты упаковочной линии и т.д., а также независимым проверкам, проводимым сотрудниками отдела контроля качества.
15 Лекарственные средства для клинических исследований должны упаковываться индивидуально для каждого больного, включенного в клиническое исследование. Основой инструкций по упаковке является заказ. В отличие от правил упаковки, принятых при серийном производстве регистрированных лекарственных средств, серия лекарственных средств для клинических исследований может быть разделена на различные упаковочные серии, которые упаковываются по отдельности в разное время.
16 Количество единиц упаковываемой продукции должно быть определено до начала операций по упаковке с учетом количества единиц, необходимых для проведения контроля качества, и архивных образцов. После окончания упаковки и маркировки необходимо подвести баланс упаковочных материалов, нерасфасованной и готовой продукции.
Инструкции по маркировке
17 Этикетки должны содержать следующую информацию:
a) наименование спонсора;
b) дозированную лекарственную форму, способ введения, количество доз, а также название/шифр лекарственного средства и активность/дозировку в случае открытого исследования;
c) номер серии и/или код, позволяющие идентифицировать содержимое и операцию по упаковке;
d) идентификационный (рандомизационный) номер испытуемого лица;
e) указания по применению;
f) надпись "Только для клинических исследований";
g) фамилию и инициалы исследователя (если они не включены в код исследований);
h) код клинического исследования, позволяющий идентифицировать исследовательский центр и исследователя;
i) условия хранения;
j) срок использования в месяцах/годах (дата истечения срока годности, срок годности или дата повторного контроля при необходимости);
k) надпись "Хранить в недоступном для детей месте", за исключением тех случаев, когда продукт предназначен только для использования в условиях стационара.
Внешняя упаковка может содержать символы или пиктограммы для пояснения некоторой информации, упомянутой выше, и требование "Возвратить пустую упаковку и неиспользованное лекарственное средство". В соответствии с заказом может быть указана дополнительная информация (например, предостережение и инструкция по обращению). Копии каждого типа этикеток должны храниться в протоколе серии.
18 На первичную упаковку должна быть нанесена информация по пункту 17, перечисления а) - k) данного приложения даже в случае, если указанная информация приведена на вторичной упаковке.
19 Если на вторичной упаковке указана вся информация по пункту 17, перечисления а) - k) данного приложения, а первичной упаковкой являются блистеры или первичная упаковка небольшого размера другого вида (например, ампулы), на которой не умещается указанная информация, то на такой упаковке, по крайней мере, должна быть приведена информация согласно пункту 17, перечисления а), с) и d) и способ введения (для ампул).
20 При продлении срока годности лекарственного средства для клинических исследований к нему должна быть прикреплена дополнительная этикетка, на которой должен быть указан новый срок годности и повторен номер серии. Новая этикетка может закрыть старый срок годности, но в целях контроля качества она не должна закрывать оригинальный номер серии. Операция перемаркировки может проводиться в исследовательском центре наблюдателем(ями) или фармацевтом этого центра в соответствии с принятыми инструкциями или условиями контракта. Эта операция должна проводиться под контролем второго лица. Свидетельство о прикреплении дополнительной этикетки должно быть включено в состав документации о проведении клинического исследования и протокол серии.
Протоколы производства и упаковки серии продукции
21 Протоколы производства и упаковки серии продукции должны содержать подробную информацию, достаточную для прослеживания всей последовательности операций, а также все существенные замечания, дающие новую информацию о продукте, позволяющие улучшить производственный процесс и подтвердить используемые методики.
Производство
Исходные материалы
22 Стабильность технологического процесса в значительной степени зависит от качества исходного сырья. Поэтому следует установить химические и физические свойства исходных материалов, внести их в спецификации и организовать их контроль. Спецификации на активные исходные материалы должны быть по возможности более полными и соответствовать современному уровню знаний. В ходе разработки лекарственного средства спецификации как на активные, так и на вспомогательные исходные материалы следует периодически пересматривать и актуализировать.
23 Для выявления причины и выдачи разрешения на отклонение в технологическом процессе необходимо иметь подробную информацию о качестве активных и вспомогательных исходных материалов.
Технологические операции
24 На стадии разработки лекарственного средства не всегда можно провести аттестацию (валидацию) методик. Вследствие этого трудно заранее установить критические технологические параметры и методы их внутрипроизводственного контроля. В этих случаях критические параметры выбираются по аналогии. Основной персонал должен уделять особое внимание составлению всех необходимых инструкций и их своевременному обновлению на основе накопленного опыта.
25 Одной из существенных составляющих контроля технологических операций является сопоставление выхода продукции. Необходимо постоянно сравнивать фактический и теоретический выходы продукции и расследовать причины любых существенных отклонений.
26 Инактивацию/удаление вирусов и/или других загрязнений биологического характера следует проводить в том же объеме, что и для зарегистрированных лекарственных средств. Инструкции по очистке должны быть очень четкими и разрабатываться с учетом недостаточности информации о токсичности лекарственного средства для клинических исследований. Если процессы (например, смешение) не прошли аттестацию (валидацию), необходимо проводить дополнительные испытания контроля качества.
Требования к препарату сравнения
27 В клинических исследованиях, когда исследуемое лекарственное средство сравнивается с зарегистрированным препаратом, необходимо обеспечить целостность и качество препарата сравнения (готовой лекарственной формы, упаковочных материалов, условий хранения и пр.). Если необходимо провести существенные изменения лекарственного препарата, объем информации о нем (например, по стабильности, сравнительной растворимости, биодоступности) должен быть достаточным для гарантии того, что эти изменения не окажут существенного влияния на исходные параметры качества этого лекарственного препарата.
28 Срок годности препарата сравнения, указанный на первоначальной упаковке, не может оставаться таким же для переупакованного препарата. Поэтому спонсор обязан определить соответствующий срок годности (руководствуясь видом препарата, характеристиками упаковки и условиями, в которых этот препарат будет храниться) и указать его на этикетке. Новый срок годности не может превышать указанный на первоначальной упаковке. Если данные о стабильности препарата отсутствуют или стабильность препарата не может быть обеспечена в ходе проведения клинического исследования, новый срок годности должен быть не более 25% периода времени между датами переупаковки и истечения прежнего срока годности препарата или 6 мес от даты переупаковки препарата, в зависимости от того, какой срок меньше.
Код рандомизации
29 В инструкциях должны быть четко описаны все операции (формирование, распространение, обращение и хранение) с любым кодом рандомизации, используемым для упакованных лекарственных средств для клинических исследований.
Слепой метод
30 Следует разработать систему, позволяющую точную идентификацию "слепых" препаратов. Эта система, наряду с кодом и списком рандомизации, должна позволять правильно идентифицировать лекарственное средство, в т.ч. любую необходимую прослеживаемость к кодам и номеру серии лекарственного средства до операции по его "слепому" разделению.
31 Образцы "слепых" лекарственных средств для клинических исследований необходимо сохранять.
Контроль качества
32 Поскольку многие процессы не могут быть стандартизованы и аттестованы (валидированы), возрастает роль проведения испытаний конечного продукта на соответствие требованиям спецификаций.
33 Контроль качества должен уделять особое внимание соответствию требованиям спецификаций, относящихся к эффективности лекарственных средств, а именно:
- точности терапевтической или разовой дозы (гомогенности, однородности дозирования);
- высвобождению активных веществ (растворимости, времени растворения и т.п.);
- оценке стабильности (при необходимости, в условиях ускоренного старения и стрессовых условиях), определению предварительных условий хранения и срока годности препарата.
При необходимости контроль качества должен включать в себя подтверждение соответствия внешнего вида, запаха и вкуса "слепых" лекарственных средств.
34 За хранение контрольных образцов каждой серии продукции несет ответственность производитель или импортер, который выпустил эту серию для использования в Российской Федерации. Образцы следует хранить в первичной упаковке, используемой при проведении исследования, или в упаковке для хранения нерасфасованного лекарственного средства. Образцы хранятся не менее одного года после окончания срока годности лекарственного средства или не менее двух лет после окончания клинических исследований, в зависимости от того, какой срок больше. Если образец хранится в упаковке, отличной от упаковки, используемой при проведении клинического исследования, то для подтверждения срока годности препарата в этой упаковке должны быть данные относительно его стабильности в этой упаковке.
Выдача разрешения на выпуск серии
35 Выдача разрешения на выпуск лекарственного средства во многих случаях проводится в два этапа (до и после его окончательной упаковки):
- при проведении оценки нерасфасованной продукции должны быть рассмотрены все существенные факторы, в т.ч. условия производства, результаты внутрипроизводственного контроля, производственная документация, соответствие требованиям досье на лекарственное средство и заказу;
- при проведении оценки готового лекарственного средства помимо всех факторов, включенных в оценку нерасфасованной продукции, следует рассматривать условия, в которых проводится упаковка, результаты внутрипроизводственного контроля, документацию по упаковке, соответствие требованиям досье на лекарственное средство и заказу.
Свободное перемещение
36 Поскольку выдача разрешений на выпуск лекарственных средств для клинических исследований ("технический зеленый свет") осуществляется персоналом соответствующей квалификации, то при наличии документального свидетельства о проведении необходимых процедур контроля и выпуска продукции не требуется проведение анализов качества этой продукции после ее отгрузки в другие государства, имеющие с Российской Федерацией соответствующие соглашения.
Контракт на производство и проведение анализов
37 Помимо прочих условий в контракте должно быть четко указано, что данное лекарственное средство предназначено для проведения клинических исследований. Необходимо тесное взаимодействие между контрагентами.
Рекламации
38 Выводы по результатам любого расследования, проведенного в связи с рекламацией, должны обсуждаться между спонсором клинического исследования и производителем (при разногласиях), или между ответственным лицом производителя и лицами, ответственными за проведение соответствующего клинического исследования, с целью оценки возможного влияния рекламации на данное исследование и разработку этого лекарственного средства.
Отзыв и возврат
39 Действия по возврату лекарственных средств для клинических исследований (например, отзыв бракованного препарата, возврат после завершения исследования, возврат препаратов с истекшим сроком годности) должны быть приведены в инструкциях, разработанных и оформленных документально. Помимо лиц(а), ответственных(ого) за отзыв препарата, с инструкциями должны быть ознакомлены спонсор клинического исследования, исследователь и наблюдатель.
Отгрузка. Возврат. Уничтожение
40 Отгрузка, возврат и уничтожение неиспользованной продукции должны выполняться в соответствии с письменными инструкциями.
Отгрузка
41 Отгрузка лекарственных средств, предназначенных для клинических исследований, выполняется в соответствии с заказами на отгрузку, предоставляемыми спонсором.
42 Лекарственное средство для клинических исследований может быть отправлено в адрес исследователя только после прохождения процедуры выпуска продукции, включающей в себя две стадии: выпуск продукции после контроля качества ("технический зеленый свет") и разрешение на использование продукции, выдаваемое спонсором ("подтверждающий зеленый свет"). Разрешения должны быть оформлены документально и сохранены.
43 Упаковка должна обеспечивать сохранность и безопасность продукции при транспортировании и промежуточном хранении. Необходимо выявлять факты вскрытия и нарушения внешней упаковки при транспортировании.
44 Спонсор должен обеспечивать отгрузку продукции надлежащего качества и доставку ее по нужному адресу.
45 Необходимо хранить опись поставок, сделанных производителем, особенно идентификации адресатов поставок.
46 Перемещение исследуемых лекарственных средств из одного исследовательского центра в другой допускается в исключительных случаях и разрешается только для очень дорогих препаратов, ограниченного количества препарата, имеющегося в наличии для проведения исследований, или в чрезвычайных ситуациях. Такие перемещения должны регулироваться инструкциями, различающимися в зависимости от первоначального места хранения перемещаемого препарата (со склада, находящегося под контролем спонсора, из аптеки исследовательского центра или от исследователя). Если перемещаемый препарат хранился непосредственно у исследователя, а не в аптеке, необходимо предпринять достаточные меры предосторожности и контроля, прежде чем использовать этот препарат в другом исследовательском центре. Как правило, для подтверждения, что препарат все еще подходит для целевого использования и новой отгрузки, его следует возвратить спонсору для перемаркировки и повторного проведения полного набора испытаний на соответствие этого препарата требованиям спецификации.
Возврат
47 Возврат лекарственных средств для клинических исследований должен проводиться на согласованных условиях, определенных спонсором и изложенных в письменных инструкциях, утвержденных руководством.
48 Возвращенную продукцию следует четко маркировать и хранить в специально предназначенных зонах. Необходимо вести и хранить протоколы инвентаризации возвращенной продукции.
Уничтожение
49 Спонсор несет ответственность за уничтожение неиспользованных лекарственных средств для клинических исследований. Не допускается уничтожение лекарственных средств для клинических исследований производителем без получения разрешения от спонсора.
50 Протоколы уничтожения продукции должны быть составлены так, чтобы можно было проследить все операции. Эти протоколы следует хранить у спонсора. Лекарственные средства для клинических исследований могут быть уничтожены только после завершения клинического исследования и составления заключительного отчета.
51 Если уничтожение продукции поручается производителю, он должен предоставить спонсору паспорт или акт об уничтожении продукции. В этих документах должны быть указаны номера серий и/или шифры больных и фактическое количество уничтоженной продукции.
Приложение 14
Производство лекарственных средств из крови или плазмы человека
Основные положения
Для биологических медицинских препаратов, получаемых из крови или плазмы человека, исходным материалом являются клетки и жидкая часть крови - плазма. Лекарственные средства, получаемые из крови или плазмы человека, обладают рядом особенностей, связанных с природой исходного материала. Например, исходный материал может содержать биологические агенты, прежде всего вирусы, распространяющие заболевания. Безопасность лекарственных средств зависит как от проверки исходного материала и его источника, так и от последующих производственных операций, в т.ч. от удаления и инактивации вирусов.
Основные требования настоящего стандарта распространяются на производство продукции на основе человеческой крови или плазмы, если не оговорено иное. На такие производства могут распространяться и требования ряда приложений (например, по производству стерильных лекарственных средств, использованию ионизирующего излучения, производству биологических медицинских препаратов, использованию систем с компьютерным управлением и контролем и т.д.).
Поскольку качество готовой продукции определяется всеми этапами производства, в т.ч. сбором крови или плазмы, все операции должны выполняться в соответствии с принятой системой обеспечения качества и требованиями настоящего стандарта.
Следует принимать меры по предотвращению передачи инфекционных заболеваний и выполнению требований соответствующих нормативных документов, относящихся к плазме для фракционирования и медицинских продуктов, полученных из крови или плазмы. Следует руководствоваться также требованиями, регламентирующими подбор доноров крови, плазмы и обследование донорской крови, а также рекомендациями по изготовлению, использованию и обеспечению качества компонентов крови.
В настоящем приложении не рассматриваются компоненты крови, применяемые в трансфузионной медицине. Однако многие из изложенных положений могут быть применены при изготовлении компонентов крови, и компетентные органы могут принимать рекомендации с учетом настоящего приложения.
Термины и определения
кровь (blood): Цельная кровь, полученная от одного донора и подготовленная для трансфузии или дальнейшей переработки.
компоненты крови (blood components): Лечебные компоненты крови (эритроциты, лейкоциты, плазма, тромбоциты), полученные центрифугированием, фильтрацией и замораживанием с использованием методологии, установленной в банке крови.
лекарственные средства, получаемые из крови или плазмы (medicinal products derived from blood or plasma): Определение этому термину дается в отдельном документе.
Управление качеством
1 Обеспечение качества распространяется на все стадии производства готового продукта от сбора исходного материала (в т.ч. отбора доноров, контейнеров для крови/плазмы, антикоагулянтов и набора реактивов для тестов) до хранения, транспортирования, процесса переработки, контроля качества и доставки готовой продукции. Все процедуры должны соответствовать требованиям настоящего приложения.
2 Кровь или плазма, используемая как исходный материал для производства медицинских препаратов, должна быть заготовлена в специальных учреждениях и проверена в утвержденных лабораториях.
3 В заготавливающей организации должны быть инструкции, определяющие пригодность каждого донора к сдаче крови и плазмы (исходного материала для производства медицинских препаратов). Результаты испытаний полученного исходного материала должны быть документально оформлены и доступны производителю лекарственных средств.
4 Система постоянного контроля качества должна прослеживать этапы изготовления медицинского препарата, полученного из крови или плазмы человека, таким образом, чтобы выявлять любые отклонения от требований к качеству.
5 Лекарственные препараты, полученные из крови и плазмы человека, возвращенные как неиспользованные, как правило, повторно потребителю не выдаются (пункт 5.65 настоящего стандарта).
Помещения и оборудование
6 Помещения, используемые для сбора крови или плазмы, должны быть достаточных размеров, удобной планировки и расположения для выполнения основных операций, санитарной обработки и обслуживания оборудования. Операции по заготовке, обработке и проведению испытаний крови и плазмы следует выполнять в различных помещениях. При собеседовании с донором должны быть выполнены условия конфиденциальности информации.
7 Конструкция оборудования для производственных процессов, сбора исходного материала и проведения испытаний должна соответствовать его целевому назначению и обеспечивать выполнение требований безопасности. Оборудование подлежит аттестации до ввода в эксплуатацию. Следует проводить регулярное техническое обследование и калибровку (поверку) оборудования с оформлением необходимой документации.
8 При изготовлении медицинских препаратов из плазмы крови предусматриваются операции инактивации или удаления вирусов. Во избежание перекрестного загрязнения необходимо четко разграничивать помещения и оборудование для материала, подвергнутого обработке по инактивации или удалению вирусов, и материала, не прошедшего такие операции.
Сбор крови и плазмы
9 Взаимоотношения между производителем медицинских препаратов из крови или плазмы человека и учреждением, заготавливающим кровь (плазму), или организацией, ответственной за сбор крови (плазмы), определяются договором, содержание которого должно соответствовать установленным требованиям.
10 Личность каждого донора следует устанавливать при приеме и проверять перед взятием крови.
11 Метод дезинфекции кожи донора должен быть четко определен и обоснован. Необходимо строго следовать принятому методу.
12 Номер отобранной порции крови на этикетке должен перепроверяться независимо разными лицами для уверенности в его идентичности, указанной на упаковке крови, образце для исследований и в записи, сделанной при сдаче крови.
13 Пластиковый мешок для крови и системы для афереза должны быть осмотрены до использования при взятии крови или плазмы для выявления повреждений или загрязнения микрофлорой. Для прослеживания движения каждой единицы крови (плазмы) следует зарегистрировать номер заводской серии пластикового мешка и системы для афереза.
Прослеживаемость крови (плазмы) и действия, выполняемые после сбора крови (плазмы)
14 Должна быть организована система, позволяющая прослеживать движение каждой единицы крови (плазмы) на всех этапах ее прохождения, начиная от донора и до применения продукта, в т.ч. до потребителя (больницы или практикующего врача). Потребитель несет ответственность за идентификацию реципиента.
15 Учреждение, заготавливающее кровь или плазму, и производитель препаратов должны информировать друг друга в следующих случаях:
- обнаружение несоответствия здоровья донора установленным критериям;
- обнаружение у донора при сдаче крови позитивного результата тестирования на маркеры вирусов при отрицательных результатах, полученных при предыдущих сдачах крови;
- выявление несоответствия проведения тестирования на наличие вирусов действующей инструкции;
- болезнь донора, вызванная переносимыми препаратами крови инфекционными агентами (HBV, HCV, HAV и другими ни-А , ни-В, ни-С вирусами гепатита, HIV 1 и 2 и другими известными в настоящее время вирусами);
- выявление у донора болезни Кротцфельд-Якоба_(Creutzfeld-Jakob) (CJ или vCJD);
- обнаружение у реципиента инфекционного заболевания, источником которого был (или мог быть) донор.
Порядок информирования и необходимые действия при возникновении перечисленных выше ситуаций регламентируются инструкцией. Следует проводить анализ предыдущих случаев сдачи крови (плазмы) за период не менее 6 мес, предшествующий последней сдаче крови с негативными результатами теста. При выявлении одного из перечисленных случаев следует повторно рассмотреть и оценить документацию на серию продукции. Требуется тщательно оценить необходимость отзыва всей серии, принимая во внимание вид заболевания, объем пула крови (плазмы), интервал времени между сдачей крови и обработкой сыворотки, характеристику продукта и технологию производства. Если установлено, что в пул включена плазма, инфицированная HIV или вирусами гепатитов А, В или С, эта информация, наряду с решением предприятия о возможности продолжения производственного процесса из этого пула крови (плазмы) или необходимости отзыва этого продукта(ов), доводится до сведения контролирующих органов.
Производство и контроль качества
16 Кровь и плазма, продукты их переработки должны испытываться с использованием аттестованных (валидированных) методов требуемой чувствительности и специфичности к следующим маркерам возбудителей трансмиссивных заболеваний:
- HBsAg;
- антитела к HIV 1 и HIV 2;
- антитела к HCV.
Если один из этих тестов дает повторный положительный результат, то порция крови (плазмы) отклоняется. В нормативных документах может быть предусмотрено проведение дополнительных тестов.
17 Температуру хранения крови, плазмы и промежуточных продуктов во время складирования и транспортирования от заготавливающих учреждений до перерабатывающих предприятий следует регламентировать. Эти условия необходимо соблюдать при доставке продукции потребителю.
18 Первый гомогенный пул плазмы крови (в т.ч. после отделения криопреципитата) должен испытываться с использованием аттестованных (валидированных) методов требуемой чувствительности и специфичности. При проведении этих тестов в пуле плазмы крови не должна выявляться положительная реакция на следующие маркеры специфических трансмиссивных инфекционных агентов:
- HBsAg;
- антитела к HIV 1 и HIV 2;
- антитела к HCV.
При позитивных результатах этот пул не должен быть допущен до производства.
19 В производственный процесс может быть допущен пул плазмы, прошедший тестирование на HCV RNA методом NAT (технология амплификации нуклеиновых кислот; nucleic acid amplification). Для тестирования должны быть использованы аттестованные (валидированные) методы достаточной чувствительности и специфичности.
20 Требования к проведению испытаний на наличие вирусов или других инфекционных агентов должны быть сформулированы с учетом знаний об опасности инфекционных агентов и пригодности применяемых методик испытания.
21 Этикетки на контейнерах с отдельными дозами плазмы, хранящимися перед объединением и фракционированием, должны соответствовать действующим требованиям. Этикетки должны содержать, по крайней мере, следующую информацию: идентификационный номер отобранной порции крови, наименование и адрес донорского центра или трансфузионной службы, ответственной за препараты, серийный номер контейнера, температуру хранения, полную массу или объем плазмы, тип антикоагулянта и дату заготовки и/или разделения крови.
22 С целью уменьшения риска микробного загрязнения плазмы или внесения инородного материала, операции по объединению и размораживанию плазмы должны выполняться в чистой зоне не ниже типа D; персонал при этом должен иметь соответствующую одежду, маски и перчатки. Необходимо регулярно контролировать методики открывания контейнеров, объединения и размораживания плазмы, например, путем проведения испытаний на наличие микробного загрязнения.
23 Следует четко разграничивать промежуточный или конечный продукты, прошедшие операции по инактивации или удалению вирусов, от материалов, не подвергнутых такой обработке.
24 Не допускается проводить аттестацию (валидацию) методов удаления или инактивации вирусов на оборудовании, используемом для производства, во избежание загрязнения продукции вирусами, применяемыми для аттестации (валидации).
Хранение образцов
25 По возможности, индивидуальные образцы крови следует сохранять для облегчения проведения в случае необходимости ретроспективного анализа. Обычно это входит в обязанность заготавливающего учреждения. Образцы каждого пула плазмы должны храниться в соответствующих условиях не менее одного года по окончания срока годности продукта, имеющего наиболее продолжительный срок хранения.
Утилизация отбракованной крови/плазмы и промежуточных продуктов
26 Необходимо наличие инструкции по безопасной и эффективной утилизации отбракованных крови/плазмы или промежуточных продуктов.
Приложение 15
Аттестация процессов и оборудования
Основные положения
1 С целью доказательства соответствия параметров критических процессов (оборудования) заданным требованиям производители лекарственных средств должны выполнять аттестацию (валидацию) процессов и оборудования, используемых при производстве лекарственных средств. Аттестация (валидация) также проводится при существенных изменениях в помещениях, оборудовании и процессах, которые могут оказать влияние на качество продукции. Для определения состава и объема работ по аттестации следует использовать подход, основанный на оценке рисков.
Планирование работ по аттестации (валидации)
2 Аттестация (валидация) проводится в плановом порядке. Основной состав работ по аттестации оформляется в виде программы аттестации (валидации) или эквивалентном ей документе.
3 Программа аттестации является итоговым документом, составленным в лаконичной, точной и ясной форме.
4 В программу аттестации должны быть включены, по крайней мере, следующие данные:
a) цель проведения аттестации;
b) организационная схема проведения аттестации;
c) перечень всех помещений, систем, оборудования и процессов, подлежащих аттестации;
d) форму ведения документации (протоколов и отчетов);
e) этапы и график выполнения работ;
f) контроль изменений;
g) ссылки на существующие документы.
5 В случае выполнения больших проектов может потребоваться разработка отдельных программ аттестации.
Документация
6 Требования к проведению аттестации (валидации) должны быть приведены в утвержденной методике. Методика должна содержать требования к критическим процессам (оборудованию) и критерии оценки.
7 В ходе проведения аттестации готовится отчет, содержащий ссылки на методику аттестации, результаты аттестации с объяснением любых отклонений и выводами, в т.ч. рекомендации по устранению недостатков. Любые изменения в методике аттестации должны быть обоснованы и документально оформлены.
8 После успешного завершения данного этапа аттестации оформляется заключение о возможности перехода к следующему этапу аттестации.
Аттестация
Аттестация проекта
9 Первым этапом в комплексе работ по аттестации новых помещений, систем или оборудования является аттестация проекта.
10 Следует показать и документально оформить соответствие проекта требованиям GMP.
Аттестация установленного оборудования
11 Аттестацию установленного оборудования следует выполнять для новых или реконструированных помещений, систем и оборудования.
12 При аттестации установленного оборудования проверяются, как минимум:
a) монтаж оборудования, трубопроводов, систем обслуживания и инструментов на соответствие чертежам и спецификации;
b) полнота и соответствие предоставляемой поставщиком документации по эксплуатации и техническому обслуживанию;
c) наличие документации по калибровке (поверке);
d) соответствие материалов и оборудования установленным требованиям (проекту).
При аттестации установленного оборудования могут быть выполнены и другие работы.
Аттестация функционирующего оборудования
13 Аттестация функционирующего оборудования выполняется после успешного завершения аттестации установленного оборудования.
14 При аттестации функционирующего оборудования выполняются, как минимум:
a) испытания и проверки, исходя из специфических особенностей процессов, систем и оборудования;
b) испытания функционирования оборудования при рабочих параметрах, равных верхним и нижним допустимым пределам, т.е. в условиях "наихудшего случая".
При аттестации функционирующего оборудования могут выполняться и другие работы.
15 После завершения аттестации оборудования в функционирующем состоянии должны быть разработаны инструкции по эксплуатации, калибровке (поверке) и очистке, проведено обучение персонала и налажена система технического обслуживания. После этого можно проводить официальную приемку помещений, систем и оборудования.
Аттестация эксплуатируемого оборудования
16 Аттестация эксплуатируемого оборудования выполняется после успешного завершения аттестации установленного и функционирующего оборудования.
17 При аттестации эксплуатируемого оборудования выполняются, как минимум:
a) испытания и проверки с использованием реальных материалов, аттестованных заменителей или имитаторов продукта с учетом специфики процессов, помещений, систем или оборудования;
b) испытания и проверки при рабочих параметрах, равных верхним и нижним допустимым пределам.
При аттестации эксплуатируемого оборудования могут выполняться и другие работы.
18 Несмотря на то, что аттестация эксплуатируемого оборудования рассматривается как отдельный вид работ, в некоторых случаях она может выполняться в сочетании с аттестацией функционирующего оборудования.
Аттестация действующих помещений, систем и оборудования
19 В процессе текущей эксплуатации следует получать данные, обосновывающие и подтверждающие соответствие рабочих критических параметров заданным требованиям. Инструкции (методики) и отчеты по эксплуатации, калибровке, очистке, техническому обслуживанию и обучению персонала должны быть документально оформлены.
Аттестация (валидация) процесса
Общие положения
20 Изложенные в настоящем разделе требования относятся к первоначальной аттестации новых процессов, последующей аттестации измененных процессов и повторной аттестации.
21 Аттестация процесса, как правило, должна быть завершена до начала реализации лекарственных средств (перспективная аттестация). В исключительных случаях, когда провести перспективную аттестацию невозможно, может возникнуть необходимость проведения аттестации процесса в ходе производства (текущая аттестация). Действовавшие в течение некоторого времени процессы также подлежат аттестации (ретроспективная валидация).
22 Следует выполнить аттестацию помещений, оборудования, процессов и аналитических методов контроля. Персонал, выполняющий аттестацию, должен пройти соответствующее обучение.
23 Следует периодически оценивать функционирование помещений, систем, оборудования и процессов с целью подтверждения их работы в соответствии с заданными требованиями.
Перспективная аттестация
24 В состав работ и документации по перспективной аттестации входят, как минимум:
a) краткое описание процесса;
b) перечень критических стадий процесса, которые подлежат исследованию;
c) перечень используемых помещений и оборудования (в т.ч. контрольное, измерительное и регистрирующее оборудование) с указанием наличия сведений об их калибровке (поверке);
d) спецификации на готовую продукцию;
e) перечень используемых аналитических методов (при необходимости);
f) предлагаемые виды внутрипроизводственного контроля и критерии приемлемости;
g) дополнительные испытания и критерии приемлемости и аттестация аналитических методов;
h) план отбора проб;
i) порядок оформления и оценки результатов;
j) обязанности и ответственность лиц, причастных к аттестации;
k) предполагаемый график выполнения работ.
Могут быть выполнены и другие работы.
25 При выполнении этих работ (в т.ч. использование специфицированных материалов) может быть произведено несколько серий готовой продукции при обычных условиях. Теоретически число производственных циклов и наблюдений должно быть достаточным, чтобы выявить тенденции изменения параметров и получить достаточные данные для оценки. Для аттестации процесса считается достаточным выполнить три последовательных серии (производственных цикла), при которых параметры находятся в заданных пределах.
26 Размер серии при аттестации должен быть равным размеру серии при промышленном выпуске продукции.
27 Если предполагается продажа или поставка серий, произведенных при аттестации, то условия их производства должны полностью соответствовать требованиям настоящего стандарта и нормативной документации, в т.ч. успешное проведение аттестации.
Текущая аттестация
28 В исключительных случаях допускается не завершать программу аттестации до начала серийного производства.
29 Решение о проведении текущей валидации должно быть обосновано, документально оформлено и утверждено лицами, имеющими на это право.
30 К документации по проведению текущей аттестации предъявляются те же требования, что и при перспективной валидации.
Ретроспективная аттестация
31 Ретроспективная аттестация может проводиться только для хорошо отлаженных процессов. Проведение ее не допускается, если в состав продукции, технологический процесс или оборудование недавно были внесены изменения.
32 Ретроспективная аттестация таких процессов основывается на предшествующих данных. При этом требуется разработка специальной методики и проведение анализа данных предшествующей эксплуатации с выводами и рекомендациями.
33 Исходными данными для проведения ретроспективной аттестации являются протоколы производства и упаковки серий продукции, контрольные карты производства, журналы проведения технического обслуживания, данные об изменении в персонале, потенциальных возможностях процесса, данные о готовой продукции, в т.ч. карты тенденций, а также результаты изучения ее стабильности при хранении.
34 Серии продукции, отобранные для проведения ретроспективной аттестации, должны представлять репрезентативную выборку для всех серий, изготовленных в течение рассматриваемого периода, в т.ч. все серии, не соответствующие спецификациям. Количество продукции должно быть достаточным, чтобы показать стабильность процесса. При проведении ретроспективной аттестации процесса для получения данных в достаточном количестве и разнообразии могут потребоваться дополнительные испытания архивных образцов.
35 Для оценки стабильности процесса при проведении ретроспективной аттестации следует выполнить анализ данных по 10-30 последовательно произведенным сериям. Может проверяться и меньшее число серий при наличии соответствующего обоснования.
Аттестация процессов очистки
36 Аттестация процессов очистки выполняется с целью подтверждения эффективности методов очистки. При этом должны быть заданы допустимые пределы на остатки продукции, моющих средств и микробное загрязнение. Эти пределы должны быть реально достижимыми и проверяемыми.
37 Для определения установленных пределов остатков веществ или загрязнений следует использовать аттестованные аналитические методы с достаточной чувствительностью.
38 Как правило, аттестацию процессов очистки следует проводить только для поверхностей, контактирующих с продуктом. Следует также принимать во внимание детали оборудования, не вступающие в контакт с продуктом. Следует проводить аттестацию длительности интервалов времени между окончанием процесса и очисткой, а также очисткой и началом следующего процесса. Следует определить методы очистки и интервалы времени между проведением очистки.
39 Для методов очистки сходных видов продукции и процессов считается приемлемым выбрать репрезентативный ряд аналогичных материалов и процессов. В таких случаях можно проводить одно аттестационное исследование для "наихудшего случая", при котором учитываются все критические факторы.
40 Для аттестации метода очистки достаточно успешного проведения трех последовательных циклов очистки.
41 Метод "проверять, пока не будет чисто" не заменяет аттестацию процесса очистки.
42 В качестве исключения при валидации очистки можно использовать материалы, которые имитируют физико-химические свойства удаляемых веществ, если последние являются токсичными или опасными.
Контроль изменений
43 Внесение изменений в исходные материалы, компоненты продукции, технологическое оборудование, параметры окружающей производственной среды (или площадки), методы производства и контроля, которые могут повлиять на качество продукта или воспроизводимость процесса, должно выполняться по специально разработанным инструкциям. Методы контроля изменений должны предусматривать получение достаточно полных данных, чтобы убедиться, что в измененном процессе будет производиться продукт требуемого качества в соответствии с документацией.
44 Все изменения, которые могут оказать влияние на качество продукции или воспроизводимость процесса, должны быть обоснованы, оформлены документально и утверждены. Следует оценить возможное влияние изменений в помещениях, системах или оборудовании на продукцию, в т.ч. проведение анализа рисков, и определить необходимость проведения и объем работ по повторной аттестации (ревалидации).
Повторная аттестация (ревалидация)
45 Следует выполнять периодическую оценку помещений, систем, оборудования и процессов (в т.ч. процессов очистки) для подтверждения их соответствия заданным требованиям. Если существенные изменения отсутствуют, то вместо повторной аттестации достаточно составить отчет, свидетельствующий о том, что помещения, системы, оборудование и процессы соответствуют установленным требованиям.
Термины и определения
Ниже приводятся термины, относящиеся к аттестации и валидации, которые не включены в раздел "Термины и определения" настоящего стандарта, но используются в данном приложении.
анализ рисков (risk analysis): Метод оценки и описания критических параметров при функционировании оборудования или процесса.
аттестация в эксплуатации (performance qualification - PQ): Документальное подтверждение того, что помещения, системы и оборудование в комплексе работают эффективно и с воспроизводимыми показателями в соответствии с промышленным регламентом, технологическими инструкциями и спецификацией на продукт.
аттестация проекта (design qualification - DQ): Документальное подтверждение того, что проект производства (в т.ч. помещения, системы и оборудование) выполнен в соответствии с заданием на проектирование, настоящим стандартом и другими нормативными документами.
аттестация процесса (process validation): Документальное подтверждение того, что процесс, выполняемый в рамках установленных параметров, протекает эффективно и с воспроизводимыми параметрами, производя лекарственное средство, удовлетворяющее всем заданным требованиям к продукции и ее качеству.
аттестация установленного оборудования (installation qualification - IQ): Документальное подтверждение того, что монтаж помещений, систем и оборудования (установленных или измененных) выполнен в соответствии с проектом и другой технической документацией.
аттестация функционирующего оборудования (operational qualification - OQ): Документальное подтверждение того, что помещения, системы и оборудование (установленные или измененные) функционирует в соответствии с предъявляемыми требованиями во всех режимах работы.
имитирующий продукт (simulated product): Материал, который по своим физическим и, по возможности, химическим характеристикам (например, вязкости, размерам частиц, рН и пр.) близок продукту, к которому относится аттестация. Во многих случаях этими характеристиками могут обладать серии плацебо (продукта, не содержащего активного ингредиента).
контроль изменений (change control): Оформленный документально порядок, согласно которому представители различных специальностей рассматривают предложенные или существующие изменения, которые могут повлиять на статус "Аттестовано" для помещений, оборудования или процессов и определяют необходимость действий, которые должны обеспечить и документально оформить поддержание системы в статусе "Аттестовано".
наихудший случай (worst case): Условия или комплекс условий, относящихся к верхним и нижним пределам параметров производственного процесса и связанных с ними факторов, определенных документацией, которые могут привести к самой высокой вероятности появления брака в процессе или продукте по сравнению с идеальными условиями. Такие условия не всегда обязательно приводят к браку в процессе или продукте.
перспективная аттестация (prospective validation): Аттестация, выполняемая до начала серийного производства продукции, предназначенной для реализации.
повторная аттестация (ревалидация) (re-validation): Повторение первичной аттестации процесса для обеспечения гарантии того, что изменения в процессе (оборудовании), выполненные в соответствии с процедурой контроля изменений, не ухудшают характеристики процесса и качество продукции.
ретроспективная аттестация (retrospective validation): Аттестация серийного процесса производства реализуемого продукта, основанная на полученных данных о производстве и контроле серий продукции.
система (system): См. Общие термины и определения.
текущая аттестация (concurrent validation): Аттестация, выполняемая во время текущего (серийного) производства продукции, предназначенной для реализации.
Приложение 16
Подтверждение соответствия серии продукции с целью ее выпуска,
выполняемое Уполномоченным лицом
1 Область применения
1.1 В настоящем приложении устанавливается порядок подтверждения соответствия, выполняемого Уполномоченным лицом, и требования к посерийному выпуску лекарственных средств в реализацию согласно действующему порядку.
1.2 Рассмотрены также случаи, когда производство серии продукции или проведение анализов разделено на несколько этапов, выполняемых в разных местах или различными производителями, а также когда серия промежуточной или нерасфасованной продукции разделяется на две и более серий готовой продукции.
Настоящее приложение распространяется и на лекарственные средства, предназначенные для клинических исследований, производство которых имеет свои особенности (приложение 13).
1.3 Настоящее приложение не охватывает все возможные случаи подтверждения соответствия и не относится к продукции, выпуск которой регламентируется специальными требованиями (такими, как препараты крови или иммунные препараты, инсулины, газы медицинского применения и лекарственные средства для детей).
1.4 Основные требования к выпуску серии продукции приводятся в лицензии на производство и регистрационном досье. Ни одно из положений настоящего приложения не может использоваться, если оно выходит за рамки установленных требований.
2 Основные положения
2.1 Каждая серия готовой продукции должна получить от Уполномоченного лица подтверждение ее соответствия установленным требованиям до выпуска в реализацию на внутреннем рынке или на экспорт.
2.2 Целью контроля за выпуском серии продукции в реализацию являются:
- обеспечение гарантии того, что производство и контроль качества серии продукции соответствуют требованиям лицензии на производство, регистрационного досье и настоящего стандарта или аналогичного стандарта другой страны, признанного эквивалентным настоящему стандарту;
- возможность оперативного нахождения Уполномоченного лица, которое выдало разрешение на реализацию серии продукции (в случаях рекламации или отзыва продукции).
3 Введение
3.1 Производство серии лекарственных средств, в т.ч. контроль качества, разделяется на стадии, которые могут выполняться в различных местах и разными производителями. Каждая стадия должна выполняться согласно соответствующим нормативным документам, требованиям настоящего стандарта и действующему законодательству Российской Федерации. Этим должно руководствоваться Уполномоченное лицо, осуществляющее процедуру подтверждения соответствия серии готовой продукции установленным требованиям перед выпуском ее в реализацию.
3.2 На практике одно Уполномоченное лицо может не знать особенностей каждого этапа производства. Уполномоченное лицо, которое подтверждает соответствие серии готовой продукции, по отдельным вопросам может опираться на заключения других Уполномоченных лиц. В подобных случаях Уполномоченное лицо должно быть заранее уверено в надежности этих заключений, исходя из личного опыта или на основании подтверждения, полученного от других Уполномоченных лиц внутри установленной системы качества.
3.3 При выполнении отдельных этапов производства в другой стране аналогичные требования к соответствию производства и проведению контроля качества предъявляются и к участнику производства в этой стране. В этом случае производство лекарственных средств также должно осуществляться в соответствии с лицензией на производство и требованиями регистрационного досье. Производитель должен иметь лицензию на осуществление своей деятельности в соответствии с законом своей страны и выполнять требования настоящего стандарта или стандарта, признанного эквивалентным ему.
4 Общие положения
4.1 Различные стадии (этапы) производства, ввоза, контроля и хранения одной и той же серии продукции могут выполняться в разных местах. Все эти места и производственные площадки должны иметь одну или раздельные лицензии на производство и осуществлять деятельность под контролем, по крайней мере, одного Уполномоченного лица, подтверждающего соответствие этой серии установленным требованиям до ее выпуска в реализацию.
4.2 Различные серии продукции могут производиться или импортироваться и выпускаться в продажу в разных странах, имеющих соглашение с Российской Федерацией о взаимном признании условий производства и реализации. При этом держатель лицензии на производство, имеющий право на выпуск серии продукции в реализацию, должен иметь в своем распоряжении точное указание адреса площадки, на которой была выпущена конкретная серия продукции, и информацию об Уполномоченном лице, ответственном за подтверждение соответствия ее качества установленным требованиям.
4.3 Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям перед выдачей разрешения на реализацию, может основывать свое решение на личном знании всех используемых в производстве помещений и процессов, опыте участвовавшего в производстве персонала и применяемой системы качества. Оно может также опираться на заключение со стороны одного или более Уполномоченных лиц о соответствии промежуточных этапов производства установленным требованиям. Заключение должно быть оформлено документально и должно ясно определять предмет подтверждения соответствия. Порядок подтверждения должен быть оформлен документально.
4.4 Подтверждение требуется в том случае, когда Уполномоченное лицо опирается на заключение другого Уполномоченного лица. Оно должно соответствовать положениям раздела 6 настоящего стандарта. Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям, должно гарантировать соблюдение договоренностей и требований к процедурам, определенным в указанном документе. Форма документа определяется сторонами (например, в виде инструкции в рамках организации или договора между различными организациями).
4.5 Указанное подтверждение должно включать в себя обязательство со стороны поставщика нерасфасованного или промежуточного продукта ставить в известность получателя(ей) продукции обо всех отклонениях, результатах, выходящих за рамки спецификаций, несоответствиях требованиям настоящего стандарта, расследованиях, рекламациях или других событиях, которые должно принимать во внимание Уполномоченное лицо, ответственное за подтверждение соответствия серии готовой продукции всем установленным требованиям.
4.6 При использовании систем с компьютерным управлением и контролем для документального оформления подтверждения соответствия и разрешения на выпуск серии продукции следует руководствоваться правилами, приведенными в приложении 11.
4.7 При наличии подтверждения соответствия серии готовой продукции, выданного Уполномоченным лицом, не требуется повторения этой процедуры в странах, имеющих с Российской Федерацией соглашение о взаимном признании результатов подобной процедуры.
4.8 Вне зависимости от конкретных мероприятий по подтверждению соответствия и выпуску продукции, всегда должна существовать процедура выявления и быстрого отзыва всей продукции, которая может представлять опасность для потребителей.
5 Проведение испытаний и выпуск в реализацию серии продукции, произведенной внутри стран, имеющих с Российской Федерацией соглашение о взаимном признании
5.1 Производство расположено в одном месте
Если все стадии (или этапы) производства и контроля выполняются в пределах одной производственной площадки, выполнение отдельных проверок может быть передано другим лицам, но Уполномоченное лицо, подтверждающее соответствие серии готовой продукции, несет персональную ответственность за достоверность результатов проверки в рамках установленной системы качества и может принимать во внимание заключения о соответствии установленным требованиям промежуточных этапов со стороны других Уполномоченных лиц, ответственных за эти стадии.
5.2 Различные стадии производства выполняются в разных местах
Если различные стадии производства серии продукции выполняются на разных производственных площадках в пределах одной организации, то Уполномоченное лицо должно отвечать за каждую стадию производства. Подтверждение соответствия серии готовой продукции установленным требованиям должно выполняться Уполномоченным лицом производителя, которое либо несет персональную ответственность за все стадии производства, либо принимает во внимание заключения о предшествующих стадиях, сделанные Уполномоченными лицами, ответственными за эти стадии.
5.3 Отдельные промежуточные стадии производства выполняются другой организацией
Одна или более промежуточных стадий производства и контроля качества могут выполняться другой организацией. Уполномоченное лицо заказчика может принимать во внимание заключение Уполномоченного лица исполнителя о соответствующей стадии, но оно несет ответственность за то, что эта работа выполняется в соответствии с договором. Подтверждение соответствия серии готовой продукции установленным требованиям должно быть выполнено Уполномоченным лицом производителя, ответственного за выпуск серии продукции на рынок.
5.4 Из серии нерасфасованной продукции в различных местах производится несколько серий готовой продукции, которые выпускаются на рынок на основании одной лицензии на право производства лекарственных средств.
5.4.1 Уполномоченное лицо держателя лицензии на право производства лекарственных средств и их peaлизацию, выпускающее серию нерасфасованной продукции, может подтверждать соответствие всей серии готовой продукции до момента выпуска ее в реализацию. В этом случае Уполномоченное лицо либо берет на себя персональную ответственность за все стадии производства, либо принимает во внимание заключения о качестве серий продукции, полученные от Уполномоченных лиц с мест выпуска серий готовой продукции.
5.4.2 Возможно также подтверждение соответствия каждой серии готовой продукции до момента выпуска ее в реализацию Уполномоченным лицом производителя, который выполнил последнюю производственную операцию, предшествующую выпуску серии готовой продукции. В этом случае оно либо берет на себя персональную ответственность за все стадии производства, либо принимает во внимание заключение о качестве серии, полученное от Уполномоченного лица с места выпуска серии нерасфасованной продукции.
5.4.3 Во всех случаях осуществления производства готовой продукции в различных местах, на разных производственных площадках, в рамках одной лицензии на право производства лекарственных средств, должно быть определено одно лицо (как правило, Уполномоченное лицо производителя серии нерасфасованной продукции), которое несет полную ответственность за выпуск всех серий готовой продукции, полученных из одной серии нерасфасованной продукции. Это лицо должно знать о любых проблемах, связанных с качеством любой серии готовой продукции, и координировать любые действия, связанные с качеством серии нерасфасованной продукции.
Номера серий нерасфасованной и произведенной из нее готовой продукции могут не совпадать, но между ними должна прослеживаться связь, оформленная документально.
5.5 Из серии нерасфасованной продукции в различных местах, на разных площадках производится несколько серий готовой продукции, которые выпускаются на основании разных лицензий на право производства лекарственных средств.
5.5.1 Уполномоченное лицо производителя готовой продукции, подтверждающее соответствие серии готовой продукции установленным требованиям, может либо взять на себя персональную ответственность за все стадии производства, либо основываться на заключении, полученном от Уполномоченного лица производителя нерасфасованной продукции.
5.5.2 Любая проблема с качеством любой серии готовой продукции, источником которой могла явиться исходная серия нерасфасованной продукции, должна быть сообщена Уполномоченному лицу, ответственному за подтверждение качества этой серии нерасфасованной продукции. После этого Уполномоченное лицо должно предпринять все необходимые действия в отношении всех серий готовой продукции, произведенных из данной серии нерасфасованной продукции. Порядок действий в этом случае должен быть предусмотрен соответствующей инструкцией.
5.6 Серия готовой продукции закупается и реализуется держателем лицензии на право производства лекарственных средств в соответствии со своей собственной лицензией на реализацию продукции. (Это происходит, например, когда организация, поставляющая препарат-дженерик, закупает готовую продукцию, соответствие которой не было подтверждено на основании лицензии производителя, и выпускает ее на основании своей собственной лицензии на право производства и реализации лекарственных средств).
В этой ситуации Уполномоченное лицо организации, закупающей продукцию, не имеющую документальных результатов подтверждения соответствия, должно подтвердить соответствие установленным требованиям этой серии готовой продукции до момента ее реализации. При этом Уполномоченное лицо закупающей организации принимает на себя ответственность за все стадии производства или основывается на заключении о качестве серии продукции Уполномоченного лица организации-поставщика.
5.7 Лаборатория контроля качества лекарственных средств и производитель лекарственных средств действуют на основании различных лицензий на право производства лекарственных средств.
Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям, может принять на себя ответственность за проведение испытаний лекарственных средств подобной лабораторией или может основываться на подтверждении необходимого качества проведения испытаний и результатов, полученных от другого Уполномоченного лица. При отсутствии такого подтверждения Уполномоченное лицо должно знать работу данной лаборатории и методики, применяемые в ней для подтверждения соответствия качества данной готовой продукции,
6 Обязанности Уполномоченного лица
6.1 Перед осуществлением процедуры подтверждения соответствия серии готовой продукции до момента ее выпуска на рынок Уполномоченное лицо должно гарантировать следующее:
a) серия готовой продукции и процесс ее производства соответствуют лицензионным условиям и регистрационному досье;
b) серия произведена в соответствии с требованиями настоящего стандарта, а для серии продукции, импортируемой из другой страны, - в соответствии с правилами производства лекарственных средств, по крайней мере, эквивалентными требованиям настоящего стандарта;
c) основные процессы производства и методы контроля аттестованы (валидированы) с учетом фактических условий производства и протоколов на серию продукции;
d) любые отклонения или запланированные изменения в технологическом процессе или контроле качества были утверждены ответственными лицами. Любые отклонения от лицензионных условий на производство лекарственных средств и регистрационного досье были согласованы с соответствующим компетентным органом;
e) проведены все необходимые проверки и испытания (в т.ч. дополнительный отбор проб, проверки и испытания, вызванные отклонениями в технологическом процессе или плановыми изменениями);
f) документация по производственному процессу и контролю качества оформлена в установленном порядке;
g) проведены аудиты в соответствии с требованиями системы обеспечения качества;
h) приняты во внимание все факторы, которые по мнению Уполномоченного лица являются существенными для качества данной серии продукции.
Уполномоченное лицо может исполнять дополнительные обязанности в соответствии с законодательством Российской Федерации или должностными инструкциями.
6.2 Уполномоченное лицо, подтверждающее соответствие промежуточной стадии производства согласно 4.3 настоящего приложения, имеет в отношении этой стадии такие же обязанности, как и Уполномоченное лицо, выпускающее серию готовой продукции (если другое не указано в документации).
6.3 Уполномоченное лицо должно поддерживать свою квалификацию на требуемом уровне с учетом изменений в системе управления качеством.
6.4 При привлечении Уполномоченного лица к подтверждению соответствия серии продукта, который он знает недостаточно (например, при освоении нового вида продукции или при переходе на другое предприятие), Уполномоченное лицо должно приобрести необходимую квалификацию, при этом может потребоваться его повторная аттестация.
Приложение 17
Выпуск по параметрам
1 Общие положения
1.1 Определение "выпуск по параметрам", используемое в данном приложении, основано на определении Европейской организации по качеству. Оно подразумевает выпуск продукции на основе данных о значениях параметров, полученных в ходе технологического процесса, и данных о соответствии производства требованиям настоящего стандарта, которые гарантируют, что продукция имеет требуемое качество.
1.2 Выпуск по параметрам должен удовлетворять основным требованиям настоящего стандарта, соответствующих приложений и изложенным ниже требованиям.
Примечание - Порядок выдачи заключения о выпуске отдельного вида продукции по параметрам определяется в соответствующих нормативных документах.
2 Выпуск по параметрам
2.1 Проведение полного комплекса проверок и контроля параметров в процессе производства может обеспечить более высокую степень соответствия готовой продукции предъявляемым требованиям по сравнению с контролем готовой продукции.
2.2 Контроль отдельных показателей готовой продукции может быть заменен контролем параметров технологического процесса при производстве продукции. Разрешение на выпуск по параметрам выдается, изменяется или изымается лицами, несущими ответственность за оценку качества продукции, совместно с компетентными лицами, осуществляющими контроль качества лекарственных средств.
3 Выпуск по параметрам для стерильной продукции
3.1 В этом разделе рассматривается выпуск по параметрам готовой продукции без проведения испытания на стерильность. Основанием для отмены испытания на стерильность могут быть только данные по аттестации (валидации) процессов стерилизации на соответствие заданным требованиям.
3.2 Испытания на стерильность позволяют обнаружить только значительные нарушения в системе обеспечения стерильности.
3.3 Выпуск по параметрам может быть разрешен только в том случае, если параметры процесса стерилизации серии продукции гарантируют, что аттестованный (валидированный) процесс стерилизации соответствует предъявляемым требованиям.
3.4 В настоящее время выпуск по параметрам может быть разрешен только для препаратов, проходящих финишную стерилизацию в их первичной упаковке.
3.5 Для выпуска по параметрам могут рассматриваться методы стерилизации, соответствующие требованиям действующей Фармакопеи.
3.6 Выпуск по параметрам не применяется при производстве новых препаратов, поскольку для получения представительных результатов об испытаниях на стерильность требуется время. В отдельных случаях результаты испытаний выпускаемых препаратов на стерильность могут распространяться и на новый препарат, если в него внесены только незначительные изменения в плане обеспечения стерильности.
3.7 При анализе рисков системы обеспечения стерильности следует обратить внимание на оценку случаев выпуска нестерильной продукции.
3.8 Предыдущий опыт работы производителя лекарственных средств должен свидетельствовать о соответствии его производства требованиям настоящего стандарта.
3.9 При оценке соответствия производства требованиям настоящего стандарта следует учитывать выявленные случаи нарушения стерильности продукции, результаты испытаний на стерильность данного препарата, а также результаты испытаний стерильности препаратов, стерилизуемых таким же или аналогичным способом.
3.10 Участок производства и стерилизации должны обслуживать квалифицированные инженер и микробиолог с опытом работы по обеспечению стерильности.
3.11 Разработка и первоначальная аттестация (валидация) продукции и технологического процесса должны гарантировать, что при соблюдении всех условий целостность и качество продукции будут сохранены.
3.12 К рассмотрению изменений должны привлекаться лица, ответственные за обеспечение стерильности, чтo должно быть учтено в системе контроля изменений.
3.13 Следует организовать систему контроля микробного загрязнения продукции перед стерилизацией.
3.14 Следует исключить возможность перепутывания продукции, прошедшей и не прошедшей стерилизацию. Такая гарантия достигается путем физического разделения продукции или с использованием электронных систем, прошедших аттестацию (валидацию).
3.15 Протоколы стерилизации должны проверяться на соответствие требованиям спецификации с привлечением не менее двух независимых исполнителей. Такой контроль может проводиться двумя сотрудниками или сотрудником и компьютерной системой, прошедшей аттестацию (валидацию).
3.16 Перед выпуском каждой серии продукции следует дополнительно подтвердить следующее:
- все плановые работы по обслуживанию и текущие проверки используемого стерилизатора выполнены;
- все ремонтные работы и изменения согласованы с микробиологом и инженером, отвечающими за процесс стерилизации;
- используемые приборы прошли калибровку (поверку);
- срок действия аттестации (валидации) стерилизатора для данной загрузки не истек.
3.17 Если получено разрешение на выпуск серии продукции по параметрам, то решения о выпуске или отбраковке серии продукции должны быть основаны на требованиях утвержденных инструкций (спецификаций). При невыполнении этих требований выпуск продукции не допускается даже при условии успешного проведения испытания на стерильность.
Термины и определения
выпуск по параметрам (parametric release): Система выпуска продукции на основе данных о значениях параметров, полученных в ходе технологического процесса, и данных о соответствии производства требованиям настоящего стандарта, которые гарантируют, что продукция обладает требуемым качеством.
система обеспечения стерильности (sterility assurance system): Комплекс мер по обеспечению стерильности продукции.
Для препаратов, подлежащих финишной стерилизации, этот комплекс мер включает в себя следующее:
a) разработку продукта;
b) получение данных об исходных материалах и технологических вспомогательных средствах (например, газов и смазочных материалов) и, по возможности, проведение их микробиологического контроля;
c) проведение контроля загрязнения в ходе технологического процесса для предотвращения попадания микроорганизмов в продукцию и их размножения. Это достигается за счет очистки и дезинфекции поверхностей, вступающих в контакт с продукцией, выполнением работ в чистых помещениях для предотвращения попадания загрязнений из воздуха, проведением технологического процесса с ограничениями во времени и, по возможности, фильтрации;
d) предотвращение перепутывания производственных потоков стерилизованной и нестерилизованной продукции;
e) сохранение целостности продукции;
f) процесс стерилизации;
g) всю систему качества, в т.ч. систему обеспечения стерильности, а именно: контроль изменений, обучение персонала, наличие письменных инструкций, контроль за выпуском продукции, плановое техническое обслуживание, анализ ошибок и предотвращение ошибок по вине человека, аттестацию (валидацию), калибровку (поверку) и т.д.
Приложение 18
Руководство по производству активных фармацевтических субстанций (АФС)
Содержание
1 Введение
1.1 Цель
1.2 Область определения
1.3 Область применения
2 Управление качеством
2.1 Общие положения
2.2 Функции и ответственность подразделений по обеспечению и контролю качества
2.3 Функции и ответственность производственных подразделений
2.4 Внутренние аудиты (самоинспекция)
2.5 Анализ качества продукции
3 Персонал
3.1 Квалификация персонала
3.2 Гигиена персонала
3.3 Консультанты
4 Здания и системы
4.1 Проектирование и строительство
4.2 Инженерные системы
4.3 Подготовка воды
4.4 Разделение зон
4.5 Освещение
4.6 Стоки и отходы
4.7 Уборка, дезинфекция и техническое обслуживание
5 Технологическое оборудование
5.1 Требования к конструкции и монтажу
5.2 Техническое обслуживание и очистка оборудования
5.3 Калибровка (поверка)
5.4 Системы с компьютерным управлением и контролем
6 Документация и протоколы
6.1 Система документации и спецификации
6.2 Протокол очистки и использования оборудования
6.3 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы для АФС
6.4 Промышленные регламенты
6.5 Протоколы на серию продукции (протоколы производства и контроля качества серии продукции)
6.6 Протоколы лабораторного контроля
6.7 Рассмотрение протоколов серии продукции
7 Работа с материалами
7.1 Общий контроль
7.2 Приемка и карантин
7.3 Отбор проб и проведение испытаний поступивших материалов
7.4 Хранение
7.5 Повторный контроль
8 Технологический и внутрипроизводственный контроль
8.1 Технологические операции
8.2 Ограничения на время выполнения операций
8.3 Внутрипроизводственный отбор проб и контроль
8.4 Смешанные серии промежуточных продуктов или АФС
8.5 Контроль загрязнений
9 Упаковка и маркировка АФС и промежуточных продуктов
9.1 Общие положения
9.2 Упаковочные материалы
9.3 Выпуск и контроль печатных материалов
9.4 Операции по упаковке и маркировке
10 Хранение и реализация
10.1 Хранение на складе
10.2 Реализация
11 Лабораторный контроль
11.1 Общий контроль
11.2 Проведение испытаний промежуточных продуктов и АФС
11.3 Аттестация (валидация) аналитических методик
11.4 Аналитические паспорта
11.5 Контроль стабильности АФС
11.6 Срок годности и дата повторного контроля
11.7 Архивные образцы
12 Аттестация (валидация)
12.1 Политика проведения аттестации (валидации)
12.2 Документация по аттестации (валидации)
12.3 Этапы аттестации (валидации)
12.4 Аттестация (валидация) процесса
12.5 Программа аттестации (валидации) процесса
12.6 Периодическая оценка аттестованных (валидированных) систем и процессов
12.7 Аттестация (валидация) методов очистки
12.8 Аттестация (валидация) аналитических методов
13 Контроль изменений
14 Отклонение и переработка материалов
14.1 Отклонение
14.2 Повторная обработка
14.3 Переработка
14.4 Регенерация материалов и растворителей
14.5 Возврат
15 Рекламации и отзывы
16 Работа по контракту (в т.ч. проведение анализов)
17 Реализация, хранение, переупаковка и перемаркировка
17.1 Область применения
17.2 Прослеживаемость реализуемых промежуточных продуктов или АФС
17.3 Управление качеством
17.4 Переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС
17.5 Стабильность
17.6 Передача информации
17.7 Работа с рекламациями и отзывами
17.8 Работа с возвратами
18 АФС, производимые путем культивирования клеток (ферментации)
18.1 Общие положения
18.2 Поддержание банка клеток и ведение протоколов
18.3 Культивирование клеток (ферментация)
18.4 Сбор, выделение и очистка
18.5 Удаление вирусов (стадии инактивации)
19 АФС, предназначенные для проведения клинических исследований
19.1 Общие положения
19.2 Качество
19.3 Оборудование
19.4 Контроль исходных материалов
19.5 Производство
19.6 Аттестация (валидация)
19.7 Изменения
19.8 Лабораторный контроль
19.9 Документация
20 Термины и определения
1 Введение
1.1 Цель
Настоящее приложение (далее - Руководство) является руководством по применению Правил GMP к производству активных фармацевтических субстанций (АФС) с целью обеспечения гарантии их качества и соответствия требованиям к чистоте.
Термин "производство" включает в себя все виды операций с АФС: приемку исходного сырья, производство, упаковку, переупаковку, маркировку, перемаркировку, контроль качества, выпуск продукции, хранение и реализацию, а также соответствующие меры контроля. Понятия "должен", "следует", применяемые в настоящем Руководстве, указывают на рекомендации, выполнение которых предполагается, за исключением случаев, когда их выполнить невозможно или они заменены альтернативными действиями, по крайней мере, с эквивалентным уровнем обеспечения качества продукции.
Руководство не затрагивает вопросов безопасности персонала, занятого в производстве, и требований по защите окружающей среды. Производитель несет ответственность за безопасность персонала и окружающей среды в соответствии с законодательством.
Настоящее Руководство не устанавливает требований, предъявляемых при регистрации (подаче заявки на регистрацию) АФС, и не заменяет требований Фармакопеи, не затрагивает полномочий соответствующих органов по установлению специфических требований к регистрационному досье АФС для выдачи разрешения на реализацию/производство или применение лекарственных средств. Следует выполнять все требования, предъявляемые к регистрационному досье АФС.
1.2 Область определения
В нормативных документах разных стран мира существуют различия в отнесении вещества к АФС. Производство всех АФС на территории Российской Федерации, а также импортируемых АФС, должно соответствовать данному Руководству.
1.3 Область применения
Настоящее Руководство устанавливает требования к производству АФС, используемых в лекарственных средствах для человека и животных (медицинских препаратах). К производству стерильных АФС оно применимо только до стадии стерилизации. Руководство не распространяется на процессы стерилизации и производство стерильных АФС в асептических условиях. Эти процессы следует выполнять согласно требованиям настоящего стандарта (в т.ч. соответствующих приложений) и других нормативных документов.
Настоящее Руководство распространяется на АФС, производимые методами химического синтеза, экстракции, культивирования клеток (ферментации), выделения из натурального сырья или любой комбинацией этих методов. Особенности производства АФС с применением методов культивирования клеток (ферментации) приведены в разделе 18 настоящего Руководства.
Данное Руководство не распространяется на производство любых вакцин, цельных клеток, цельной крови и плазмы, производных крови и плазмы (фракционирование плазмы), а также АФС для генной терапии, но распространяется на производство АФС, получаемых с использованием крови или плазмы в качестве исходного сырья. На производство клеточных субстратов (млекопитающих, растений, насекомых или бактерий, тканей или органов животных, в т.ч. трансгенных животных) и ранние стадии производства могут распространяться требования настоящего стандарта, выходящие за рамки данного приложения. Настоящее Руководство не распространяется на производство медицинских газов, нерасфасованных лекарственных средств, а также на производство и контроль качества радиофармацевтических препаратов. Раздел 19 Руководства содержит требования, распространяющиеся только на производство АФС, используемых при получении лекарственных средств, предназначенных для клинических исследований (исследовательских лекарственных средств).
Исходный материал для производства АФС - сырье, промежуточные продукты или другие АФС, используемые в производстве АФС и включаемые в АФС в качестве существенного структурного элемента. Исходный материал для производства АФС может быть продуктом (материалом), получаемым от одного или более поставщиков по контракту (коммерческому соглашению), или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
Производитель АФС должен определить и оформить документально стадию, с которой начинается производство АФС из исходного сырья. Для процессов химического синтеза эта стадия определяется как стадия ввода исходных материалов в технологический процесс производства АФС. Для других процессов (ферментации, экстракции, очистки и пр.) такая стадия определяется с учетом конкретных особенностей производства. В таблице 1 приведены стадии производства различных АФС с выделением стадий, на которых исходный материал, как правило, вводится в технологический процесс.
Таблица 1 - Применение Руководства к производству АФС
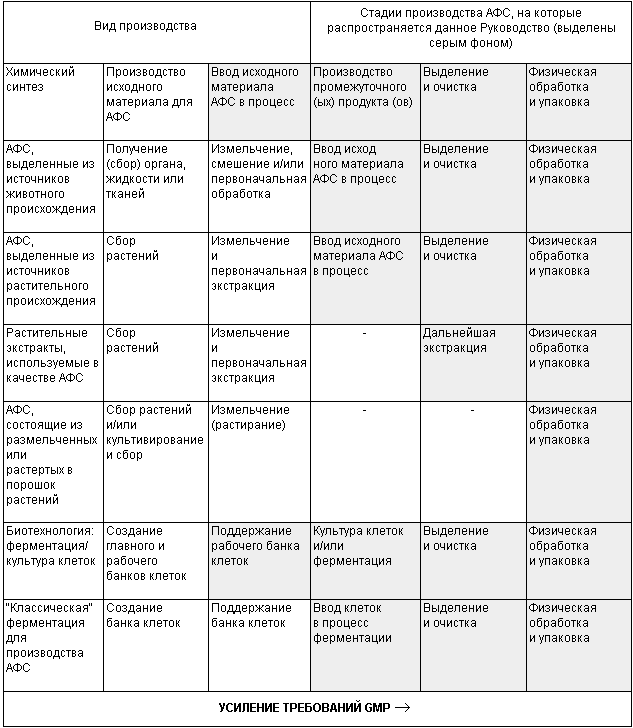
Начиная с этой стадии, на данные промежуточные продукты и/или стадии производства АФС действуют правила данного приложения. Они включают в себя требования к аттестации (валидации) критических стадий( )технологического процесса, оказывающих влияние на качество АФС. В то же время выбор стадий технологического процесса для проведения аттестации (валидации) не обязательно означает, что эти стадии являются критическими для качества АФС.
Требования настоящего приложения распространяются, как правило, на стадии, отмеченные в таблице1 серым фоном. Это не означает, что в процессе производства должны выполняться все стадии, указанные в данной таблице. Строгость следования требованиям GMP должна возрастать от ранних стадий производства АФС к завершающим стадиям технологического процесса, очистки и упаковки. Физическую обработку АФС, такую как грануляция, покрытие оболочкой или физическое изменение размера частиц (например, грубый и тонкий помол), следует проводить, по крайней мере, в соответствии с требованиями настоящего стандарта.
Настоящий стандарт не распространяется на технологические стадии, предшествующие вводу исходных материалов для производства АФС в технологический процесс.
2 Управление качеством
2.1 Общие положения
2.10 За качество выпускаемого продукта несет ответственность весь персонал предприятия-производителя.
2.11 На каждом предприятии следует разработать, документально оформить и внедрить систему управления качеством, которая предусматривает активное участие в ней руководителей предприятия и всего персонала, занятого в производстве.
2.12 Система управления качеством охватывает организационную структуру, инструкции, методики, процессы и ресурсы, а также действия, необходимые для обеспечения гарантии соответствия АФС всем требованиям к качеству и чистоте. Деятельность, связанная с обеспечением качества, должна быть четко определена и оформлена документально.
2.13 Подразделения (отделы, службы), ответственные за обеспечение и контроль качества, должны быть независимыми от производства. Эти функции могут выполняться отдельными подразделениями по обеспечению и контролю качества либо быть возложены на одно лицо (группу лиц) в зависимости от объемов и структуры предприятия.
2.14 Следует определить круг лиц, ответственных за выпуск промежуточных продуктов и АФС.
2.15 Все действия по контролю качества следует оформлять документально непосредственно при их выполнении.
2.16 Любые отклонения от принятых инструкций должны быть обоснованы и оформлены документально. Критические отклонения следует расследовать с необходимым документальным оформлением.
2.17 Не допускается выпускать или использовать материалы до получения положительного заключения отдела(ов) качества, если не установлен специальный порядок, допускающий соответствующие отклонения (например, выпуск материалов, находящихся на карантине, приведенных в пункте 10.20 настоящего Руководства, или использование сырья или промежуточных продуктов, оценка качества которых еще не завершена).
2.18 Следует разработать порядок своевременного извещения руководства предприятия о результатах инспекций, проводимых органами контроля и надзора, случаях серьезного нарушения требований настоящего стандарта, обнаружения несоответствия продукта и принятых мерах (претензиях к качеству продукции, отзывах продукции, заключениях инспекции и т.д.).
2.2 Функции и ответственность подразделений по обеспечению и контролю качества
2.20 Деятельность подразделений по обеспечению и контролю качества распространяется на все аспекты качества.
2.21 Подразделения по обеспечению и контролю качества должны рассматривать и согласовывать (утверждать) все документы, имеющие отношение к качеству продукции.
2.22 Функции и ответственность службы качества, независимой от производства, не могут быть переданы другим подразделениям или лицам. Функции и ответственность этой службы должны быть оформлены документально и включать в себя, по крайней мере, следующее:
1 Выпуск или отзыв любых АФС. Выпуск или отзыв промежуточных продуктов, используемых вне зоны контроля предприятия-производителя.
2 Разработку и ввод в действие системы выпуска или отзыва сырья, промежуточных продуктов, упаковочных и печатных материалов (материалов для маркировки).
3 Проверку серии готовой продукции и протоколов лабораторных анализов на критических стадиях технологического процесса до выпуска АФС в реализацию.
4 Расследование причин критических отклонений параметров от установленных значений с принятием необходимых мер и т.д.
5 Согласование всех спецификаций и технологических инструкций по производству продукции.
6 Согласование всех инструкций и методик по качеству промежуточных продуктов или АФС.
7 Контроль проведения внутренних проверок (самоинспекций).
8 Согласование производителей промежуточных продуктов и АФС, работающих по контракту.
9 Согласование изменений, которые могут оказать влияние на промежуточные продукты и АФС.
10 Рассмотрение и согласование протоколов и отчетов о проведении аттестации (валидации).
11 Контроль за расследованием претензий, связанных с качеством продукции, и принятием необходимых мер.
12 Контроль своевременного выполнения технического обслуживания и калибровки (поверки) критического оборудования в соответствии с установленным порядком.
13 Контроль проведения испытаний и оформления их результатов.
14 Контроль (при необходимости) данных о стабильности АФС и/или промежуточных продуктов для обоснования даты их повторного контроля, срока годности и условий их хранения.
15 Составление отчетов о качестве продукции (подраздел 2.5 настоящего Руководства).
2.3 Функции и ответственность производственных подразделений
Функции и ответственность производственных подразделений должны быть оформлены документально и включать в себя, по крайней мере, следующее:
1 Разработку, пересмотр и согласование (утверждение) инструкций по производству промежуточных продуктов и/или АФС, а также обеспечение инструкциями всех причастных подразделений и исполнителей в cooтветствии с утвержденной инструкцией.
2 Производство АФС и, при необходимости, промежуточных продуктов в соответствии с утвержденными инструкциями.
3 Рассмотрение всех протоколов производства серии продукции и подтверждение их полноты и оформления (подписания) в установленном порядке.
4 Контроль проведения анализа всех отклонений в ходе производства и расследований критических отклонений с соответствующим документальным оформлением.
5 Контроль чистоты производственных помещений и, при необходимости, их дезинфекции.
6 Контроль выполнения необходимых калибровок (поверок) и хранения протоколов калибровки.
7 Контроль содержания помещений и оборудования в надлежащем состоянии и хранения соответствующих протоколов.
8 Контроль ведения и согласования (утверждения) протоколов и отчетов аттестации (валидации) оборудования и процессов.
9 Оценка предлагаемых изменений в продукции, технологическом процессе или оборудовании.
10 Контроль проведения аттестации новых и, при необходимости, модернизированных помещений и оборудования.
2.4 Внутренние аудиты (самоинспекция)
2.40 Для подтверждения соответствия производства АФС требованиям настоящего стандарта следует регулярно проводить внутренние аудиты согласно утвержденному графику.
2.41 Результаты аудита и последующие корректирующие действия следует оформлять документально и представлять руководству предприятия. Действия по результатам аудита следует выполнять своевременно и надлежащим образом.
2.5 Анализ качества продукции
2.50 Для подтверждения постоянного соответствия технологического процесса установленным требованиям следует регулярно проводить анализ качества АФС. Такой анализ следует проводить, как правило, ежегодно с последующим документальным оформлением. Отчет о проведении анализа качества продукции должен включать в себя, по крайней мере, следующее:
- рассмотрение результатов внутрипроизводственного контроля и испытаний АФС по критическим параметрам;
- анализ данных о всех сериях продукции, качество которых не удовлетворяло установленным требованиям;
- анализ всех существенных отклонений в технологическом процессе и результаты расследования их причин;
- рассмотрение всех изменений в технологическом процессе или аналитических методах;
- рассмотрение результатов непрерывного контроля стабильности;
- анализ всех претензий, возвратов и отзывов, связанных с качеством продукции;
- анализ эффективности всех корректирующих действий, принятых по результатам аудита.
2.51 Следует оценить результаты анализа и принять решение о необходимости принятия соответствующих мер или повторной аттестации (валидации). Обоснование необходимости этих мер должно быть оформлено документально. Указанные меры должны быть выполнены своевременно и надлежащим образом.
3 Персонал
3.1 Квалификация персонала
3.10 Производство должно быть укомплектовано необходимым числом сотрудников, имеющих соответствующее образование, прошедших обучение и/или имеющих достаточный опыт для работы в производстве АФС и промежуточных продуктов.
3.11 Ответственность и обязанности всех сотрудников, работающих в производстве АФС и промежуточных продуктов, должны быть документально оформлены.
3.12 Следует проводить регулярное обучение персонала с привлечением специалистов необходимой квалификации. Это обучение должно охватывать, как минимум, вопросы, связанные с выполнением конкретных операций и требований настоящего стандарта в части функций данного сотрудника. Следует вести и сохранять протоколы обучения сотрудников, а также периодически оценивать их квалификацию.
3.2 Гигиена персонала
3.20 Персонал должен соблюдать правила гигиены.
3.21 Персонал должен носить чистую одежду, соответствующую его производственной деятельности. Следует определить порядок замены этой одежды. Для защиты АФС и промежуточных материалов от загрязнения, при необходимости, следует носить дополнительные элементы одежды, покрывающие голову, лицо и руки.
3.22 Следует избегать прямого контакта персонала с промежуточными продуктами и АФС.
3.23 Курение, прием пищи и напитков, жевание резинки и хранение продуктов питания допускаются только в специально выделенных местах, отделенных от производственных помещений.
3.24 Сотрудники с инфекционными заболеваниями или имеющие открытые повреждения на незакрытых участках тела не допускаются к работе, если это может отразиться на качестве АФС. Любой сотрудник с заболеванием или наличием открытой раны, выявленными при медицинском осмотре или наблюдении, должен быть отстранен от производственных операций, при выполнении которых состояние его здоровья может неблагоприятно повлиять на качество АФС, до выздоровления или получения медицинского заключения о том, что нахождение этого сотрудника на рабочем месте не представляет риска для безопасности или качества АФС.
3.3 Консультанты
3.30 Консультанты по вопросам производства и контроля качества промежуточных материалов или АФС должны иметь соответствующее образование, подготовку и опыт работы или сочетание вышеперечисленных навыков.
3.31 Следует организовать ведение и хранение протоколов о проведении консультаций с указанием имен, адресов, специальностей и видов услуг, предоставляемых этими консультантами.
4 Здания и системы
4.1 Проектирование и строительство
4.10 При размещении, проектировании и строительстве зданий и помещений, предназначенных для производства промежуточных продуктов и АФС, следует предусматривать удобство их эксплуатации и обслуживания в соответствии с видом и стадией производства. При проектировании помещений следует сводить к минимуму возможность загрязнения. Если для промежуточных продуктов и АФС устанавливаются требования по микробной чистоте, то при проектировании помещений следует предусматривать защиту от нежелательных микробных загрязнений.
4.11 Здания и помещения должны быть достаточных размеров для размещения оборудования и материалов с целью предотвращения их перепутывания и загрязнения.
4.12 Оборудование, обеспечивающее надлежащую защиту материалов (например, закрытая или изолированная система), может частично выходить за пределы производственного помещения.
4.13 При организации потоков материалов и персонала в зданиях или помещениях следует предусматривать предотвращение перепутывания и загрязнения материалов и продукции.
4.14 Следует предусматривать отдельные зоны или другие адекватные решения для следующих операций:
- получения, идентификации, отбора проб и нахождения в карантине исходных материалов до выдачи разрешения на выпуск или отклонение;
- нахождения в карантине до выпуска или отклонения промежуточных продуктов или АФС;
- отбора проб промежуточных продуктов или АФС;
- хранения отклоненных материалов до принятия решения об их дальнейшем использовании (например, при возврате, переработке или уничтожении);
- хранения выпущенных материалов;
- выполнения технологических операций;
- выполнения операций по упаковке и маркировке;
- проведения лабораторных анализов.
4.15 Следует предусматривать необходимые помещения для подготовки персонала (мытье рук и пр.) и туалеты, обеспечить их горячей и холодной водой, мылом или другими моющими средствами, фенами для рук или одноразовыми полотенцами. Помещения для подготовки персонала и туалеты должны быть отделены от производственных зон (не иметь в них прямого выхода), но должны быть легкодоступными для персонала. При необходимости следует предусмотреть помещения для принятия душа и/или переодевания.
4.16 Как правило, лаборатории и помещения, в которых проводятся анализы, должны быть расположены отдельно от производственных зон. Если проводимые в зонах технологические операции не влияют на результаты анализа, а работа лабораторий не оказывает отрицательного влияния на производство промежуточных продуктов и АФС, лаборатории (в частности, лаборатории внутрипроизводственного контроля) могут быть расположены в производственных зонах.
4.2 Инженерные системы
4.20 Следует выполнять аттестацию инженерного оборудования и технологических сред (например, пара, газов, сжатого воздуха, систем отопления, вентиляции, кондиционирования и пр.), которые могут оказать влияние на качество продукции, и обеспечить их контроль и техническое обслуживание. При несоответствии параметров заданным значениям следует принимать необходимые меры. На предприятии должен храниться соответствующий комплект документации.
4.21 При необходимости следует предусматривать системы вентиляции, очистки и вытяжки воздуха. При проектировании и мониторинге этих систем следует свести к минимуму риск прямого и перекрестного загрязнения и предусмотреть соответствующий контроль параметров для каждой технологической стадии (например, давления воздуха, наличия микроорганизмов (при необходимости), запыленности, влажности и температуры). Особое внимание следует уделить зонам, в которых АФС подвергаются воздействию окружающей среды.
4.22 В производственных помещениях с рециркуляцией воздуха следует предусмотреть меры по предотвращению риска прямого и перекрестного загрязнения.
4.23 Следует предусмотреть маркировку или иной метод идентификации стационарных систем трубопроводов (например, путем маркировки отдельных трубопроводов, обеспечением документации, применением систем с компьютерным управлением и контролем и пр.). Расположение системы трубопроводов не должно представлять опасности загрязнения промежуточных продуктов и АФС.
4.24 Канализационные трубы должны иметь соответствующие размеры и, при необходимости, иметь разрыв струи либо другие средства предотвращения обратного потока.
4.3 Подготовка воды
4.30 Вода, используемая при производстве АФС, должна удовлетворять своему назначению с соответствующим подтверждением.
4.31 За исключением отдельно оговоренных случаев, качество воды, используемой в производстве, должно, как минимум, соответствовать требованиям действующих стандартов, предъявляемым к питьевой воде.
4.32 Если для обеспечения качества АФС характеристик питьевой воды недостаточно и необходимы более жесткие требования к химическим и/или микробиологическим характеристикам воды, должны быть разработаны требования к воде по физико-химическим свойствам, общему числу микроорганизмов, числу нежелательных микроорганизмов и/или содержанию эндотоксинов в воде.
4.33 Следует выполнять аттестацию процесса подготовки воды определенного качества, используемой в технологическом процессе, и контролировать ее параметры с установлением необходимых пределов действия.
4.34 Если нестерильные АФС предназначены для дальнейшего производства стерильных лекарственных средств (или их производитель заявляет о пригодности АФС для этой цели), то вода, применяемая на конечных стадиях выделения и очистки, должна контролироваться на общее число микроорганизмов, число нежелательных микроорганизмов и уровень эндотоксинов.
4.4 Разделение зон
4.40 Для производства веществ с высокой сенсибилизирующей активностью (пенициллины или цефалоспорины и пр.) следует использовать специально выделенные производственные зоны, в состав которых могут входить помещения, вентиляционное и/или технологическое оборудование.
4.41 Специальные производственные площади должны быть выделены для производства веществ с инфицирующими свойствами, высокой фармакологической активностью или токсичностью (например, некоторых стероидов, цитотоксических противоопухолевых средств), за исключением случаев, когда методы инактивации и/или очистки оборудования прошли аттестацию (валидацию) и выполняются в установленном порядке.
4.42 Для предотвращения перекрестного загрязнения при перемещении персонала, материалов и т.д. из одной зоны в другую следует предусмотреть соответствующие меры.
4.43 Не допускается проводить в зданиях и/или на оборудовании, предназначенном для производства АФС, любые производственные операции (в т.ч. взвешивание, размол или упаковку) с высокотоксичными нефармацевтическими материалами, например, гербицидами и пестицидами. Обращение с высокотоксичными нефармацевтическими материалами и их хранение должны быть отделены от АФС.
4.5 Освещение
4.50 Для облегчения правильной эксплуатации, обслуживания и очистки помещений следует предусматривать необходимый уровень освещенности.
4.6 Стоки и отходы
4.60 Стоки и отходы (например, твердые, жидкие или газообразные побочные продукты производства) должны своевременно и безопасно удаляться из зданий и окружающей их зоны с соблюдением санитарных норм. Контейнеры и/или трубопроводы для отходов должны иметь четкую маркировку.
4.7 Уборка, дезинфекция и техническое обслуживание
4.70 Здания и помещения, в которых производятся промежуточные продукты и АФС, должны соответствующим образом обслуживаться, ремонтироваться и содержаться в чистоте.
4.71 Для проведения уборки (обработки, дезинфекции) должны быть разработаны специальные инструкции с указанием используемого оборудования, материалов и графиков выполнения работ.
4.72 При необходимости для предотвращения загрязнения оборудования, сырья, упаковочных и печатных материалов, промежуточных продуктов и АФС следует разработать инструкции по борьбе с грызунами, использованию инсектицидов, фунгицидов, фумигирующих средств, а также чистящих и дезинфицирующих средств.
5 Технологическое оборудование
5.1 Требования к конструкции и монтажу
5.10 Оборудование, применяемое в производстве промежуточных продуктов и АФС, должно иметь соответствующую конструкцию и размеры и располагаться в месте, удобном для его использования, очистки, дезинфекции и технического обслуживания.
5.11 Поверхности оборудования, контактирующие с сырьем, промежуточными продуктами или АФС, не должны влиять на качество промежуточных продуктов и АФС в пределах установленных требований.
5.12 Технологическое оборудование должно использоваться только по назначению.
5.13 Основное оборудование и стационарные технологические линии (реакторы, контейнеры для хранения, технологические линии и пр.), используемые в производстве промежуточных продуктов и АФС, должны иметь соответствующую маркировку.
5.14 Не допускается контакт всех веществ, применяемых в технологическом оборудовании (смазок, нагревающих и охлаждающих жидкостей) с промежуточными продуктами и АФС, который может привести к изменению качества промежуточных продуктов и АФС за пределы установленных требований. Необходимо расследовать любые отклонения от этих требований, чтобы не допускать отрицательного влияния вспомогательных веществ на перерабатываемые материалы. По возможности следует использовать смазки и масла, предназначенные для пищевой промышленности.
5.15 Оборудование, по возможности, должно быть закрытым или герметичным. При использовании открытого оборудования необходимо принимать меры по снижению риска загрязнения до минимума.
5.16 Следует хранить действующий комплект документации на оборудование и установки (например, контрольно-измерительные приборы, вспомогательные системы и пр.).
5.2 Техническое обслуживание и очистка оборудования
5.20 Техническое обслуживание оборудования следует проводить в соответствии с утвержденными графиками и инструкциями, в которых должны быть указаны ответственные лица.
5.21 Инструкции по очистке оборудования и допуску его к последующему использованию в производстве промежуточных продуктов и АФС должны быть достаточно подробными и содержать следующие данные:
- определение ответственности за очистку оборудования;
- график проведения очистки и, при необходимости, дезинфекции;
- подробное описание методов и материалов, в т.ч. приготовление моющих средств;
- инструкции по разборке и сборке каждого узла оборудования (при необходимости);
- порядок удаления и уничтожения данных о предыдущей серии продукции;
- инструкции по предохранению чистого оборудования от загрязнения до его использования;
- порядок проверки чистоты оборудования непосредственно перед его использованием (при необходимости);
- максимально допустимое время от окончания производства до очистки оборудования (при необходимости).
5.22 Для предотвращения прямого и перекрестного загрязнения или переноса материалов, которые могут повлиять на соответствие качества промежуточных продуктов и АФС установленным требованиям, следует предусматривать надлежащее хранение, очистку оборудования и принадлежностей (при необходимости, дезинфекцию или стерилизацию).
5.23 Для оборудования, предназначенного для непрерывного производства или производства циклами серий одних и тех же промежуточных продуктов или АФС, следует предусмотреть очистку через определенные интервалы времени для предотвращения накопления и переноса загрязнений (например, продуктов распада или недопустимой концентрации микроорганизмов).
5.24 Для неспециализированного оборудования между производством различных материалов следует предусмотреть очистку с целью предупреждения перекрестного загрязнения.
5.25 Должны быть установлены допустимые пределы наличия остатков, методы и материалы для очистки.
5.26 Проведение очистки оборудования должно быть документально оформлено. Оборудование должно быть промаркировано.
5.3 Калибровка (поверка)
5.30 Калибровку (поверку) контрольно-измерительного и аналитического оборудования (в т.ч. весов), имеющего критическое значение для обеспечения качества промежуточных продуктов или АФС, следует проводить в плановом порядке.
5.31 Калибровку (поверку) оборудования следует выполнять с использованием стандартов (эталонов), прослеживаемых до соответствующих эталонных образцов (если они существуют).
5.32 Протоколы проведения калибровок (поверок) должны сохраняться.
5.33 Данные о состоянии калибровки (поверки) критического оборудования должны находиться в доступном месте.
5.34 Не допускается использовать приборы, не соответствующие требованиям калибровки (поверки).
5.35 При обнаружении отклонений в контрольно-измерительном оборудовании от установленных требований следует выяснить, могут ли они оказать влияние на качество промежуточных продуктов или АФС, произведенных с использованием этого оборудования после последнего проведения калибровки (поверки).
5.4 Системы с компьютерным управлением и контролем
5.40 Системы с компьютерным управлением и контролем должны быть аттестованы (валидированы) с учетом разнообразия, сложности и критичности использования компьютеров.
5.41 Пригодность систем с компьютерным управлением и контролем и программного обеспечения для выполнения поставленной задачи должна быть показана при их аттестации (валидации) в установленном и функционирующем состояниях.
5.42 Если программное обеспечение аттестовано до его получения, то не требуется проводить его проверку в аналогичном объеме. Если система с компьютерным управлением и контролем не была аттестована (валидирована) при монтаже, то при наличии необходимой документации может быть проведена ее ретроспективная аттестация (валидация).
5.43 Следует предусмотреть защиту систем с компьютерным управлением и контролем от несанкционированного доступа или изменения данных, а также защиту от потери данных (например, при выключении компьютера). Должна регистрироваться информация о любых изменениях данных, последнем вводе данных, о том, кем и когда они были сделаны.
5.44 Работа и обслуживание систем с компьютерным управлением и контролем должны проводиться в соответствии с инструкциями.
5.45 При вводе существенной информации вручную следует предусмотреть дополнительную проверку правильности ввода данных. Такая проверка может выполняться другим оператором либо самой системой.
5.46 Сбои в работе систем с компьютерным управлением и контролем, которые могут оказать влияние на качество промежуточных продуктов или АФС, на надежность записей или результатов испытаний, следует регистрировать и расследовать.
5.47 Изменения в системе с компьютерным управлением и контролем должны проводиться в соответствии с методикой внесения изменений, оформляться документально и утверждаться ответственным лицом с проверкой внесения этих изменений. Следует хранить протоколы всех изменений и усовершенствований, внесенных в компьютер, программное обеспечение или любой другой критический элемент системы с компьютерным управлением и контролем.
5.48 Если сбои или отказы в работе системы могут привести к необратимой потере данных, следует предусмотреть резервную систему. Для всех систем с компьютерным управлением и контролем должны быть предусмотрены меры, гарантирующие защиту данных.
5.49 Допускается запись информации другими средствами.
6 Документация и протоколы
6.1 Система документации и спецификации
6.10 Разработку, пересмотр, утверждение и распространение всех документов, относящихся к производству промежуточных продуктов или АФС, следует выполнять по специальным инструкциям (методикам). Эти документы могут быть представлены как в письменном, так и в электронном виде.
6.11 Выпуск, пересмотр, замена и отмена всех документов должны контролироваться с сохранением сведений об их предыдущих версиях.
6.12 Следует организовать систему хранения всех необходимых документов (отчетов о развитии производства, объемах производства, замене оборудования и аттестации технологических процессов, протоколов обучения персонала, выпуска продукции, контроля качества и реализации продукции и пр.) и установить сроки хранения этих документов.
6.13 Вся документация о производстве, контроле качества и реализации продукции должна храниться не менее одного года после окончания срока годности данной серии. Для АФС с установленной датой повторного контроля срок хранения документов - не менее трех лет со дня полной реализации серии.
6.14 Записи в протоколы следует вносить в предназначенные для этого места сразу после выполнения действий с указанием ответственного лица, сделавшего эти записи, так, чтобы их нельзя было удалить. Исправления в записях должны быть внесены таким образом, чтобы можно было прочесть первоначальную запись. Исправления должны содержать дату внесения изменений и подпись ответственного лица.
6.15 Оригиналы или копии протоколов, регистрирующие какие-либо действия, следует хранить в соответствующих подразделениях в течение всего срока хранения документации. Допускается хранить протоколы в других местах, если используется электронный или любой другой способ переноса информации.
6.16 Спецификации, инструкции, методики и протоколы могут храниться в виде оригиналов или заверенных копий (например, фотокопии, микрофильмы, микрофиши или другие точные воспроизведения оригинальных документов). При использовании микрофильмирования или записи в электронном виде следует иметь считывающее оборудование и средства для выполнения копий на жестких носителях.
6.17 Спецификации на сырье, промежуточные продукты (при необходимости), АФС, печатные и упаковочные материалы должны быть оформлены в письменном виде. Могут потребоваться спецификации для других материалов (технологических добавок, смазок и т.д.), используемых при производстве промежуточных продуктов или АФС, оказывающих решающее влияние на качество продукции. Критерии приемлемости для внутрипроизводственного контроля также должны быть оформлены в письменном виде.
6.18 Электронные подписи на документах должны быть идентифицированы и защищены.
6.2 Протокол очистки и использования оборудования
6.20 В протоколах использования, очистки, дезинфекции и/или стерилизации и обслуживания основного оборудования должны быть указаны дата, время, наименование и номер каждой серии произведенной на нем продукции, а также информация о лице, проводившем очистку и обслуживание этого оборудования.
6.21 Не требуется составление отдельных протоколов очистки и использования оборудования в случае, если оно специально предназначено для производства одного промежуточного продукта или АФС и серии этого промежуточного продукта или АФС производятся в прослеживаемой последовательности. При использовании оборудования специального назначения протоколы очистки, обслуживания и использования могут быть частью протокола на серию продукции или составлены отдельно.
6.3 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы для АФС
6.30 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы, используемые для производства АФС, должны храниться в установленном порядке и содержать следующие данные:
- наименование производителя, содержание и количество каждой поставки каждой партии сырья, промежуточных продуктов или печатных и упаковочных материалов для производства АФС, наименование поставщика, шифр(ы) или другой идентификационный номер поставщика (если известны), номер, присвоенный при поступлении и дата поступления;
- результаты проведенных испытаний или исследований и выводы по ним;
- протоколы, прослеживающие использование материалов;
- протокол о соответствии материалов, используемых для маркировки и упаковки АФС, требованиям, установленным в спецификациях, и
- принятое решение относительно отклоненного сырья, промежуточных продуктов или печатных и упаковочных материалов, используемых в производстве АФС.
6.31 Следует хранить утвержденные образцы этикеток для сравнения с ними выпускаемых этикеток.
6.4 Промышленные регламенты
6.40 С целью обеспечения однообразия и повторяемости от серии к серии для каждого промежуточного продукта и АФС должны быть разработаны промышленные регламенты, подписанные одним лицом с указанием даты, проверенные и подписанные другим независимым лицом со стороны отдела(ов) качества с указанием даты.
6.41 Промышленные регламенты должны содержать следующие данные:
- наименование производимого промежуточного продукта или АФС и ссылку на соответствующий документ (если это возможно);
- полный перечень сырья и промежуточных продуктов согласно их наименованиям или кодам, достаточным для идентификации всех их показателей качества;
- количество или соотношение используемых видов сырья или промежуточного продукта, в т.ч. единица его измерения. Если это количество не является фиксированной величиной, то в протокол должен быть включен расчет для каждого размера серии или производительности. При наличии обоснования в протокол следует включать допустимые отклонения значений;
- место производства и основное оборудование;
- подробные технологические инструкции, включающие в себя:
- последовательность операций;
- допустимые пределы изменения технологических параметров;
- инструкции по отбору проб и проведению внутрипроизводственного контроля с критериями приемлемости (при необходимости);
- ограничения во времени для завершения отдельных технологических стадий и/или всего технологического процесса;
- предполагаемый выход продукта (с указанием наибольшего и наименьшего значений) для соответствующих стадий технологического процесса или отрезков времени;
- особые указания и меры предосторожности или ссылки на них (при необходимости);
- инструкции по хранению промежуточного продукта или АФС, печатных и упаковочных материалов и особых условий хранения с указанием сроков годности.
6.5 Протоколы на серию продукции (протоколы производства и контроля качества серии продукции)
6.50 Для каждого промежуточного продукта и АФС составляются протоколы серии продукции, включающие в себя полную информацию о производстве и контроле качества каждой серии. До выпуска продукции протокол на серию должен быть проверен с целью подтверждения содержащихся в нем данных и их соответствия промышленному регламенту. Если протокол на серию продукции относится к части промышленного регламента, в протоколе должна быть ссылка на соответствующий промышленный регламент.
6.51 Протоколы на серию продукции должны быть пронумерованы с использованием уникального номера серии или идентификационного кода и подписаны с указанием даты. При непрерывном производстве до присвоения окончательного номера серии в качестве идентификационного кода можно использовать шифр продукта с указанием даты и времени.
6.52 В протоколах на серию продукции при завершении каждой важной технологической стадии следует указывать:
- дату и время, при необходимости;
- основное используемое оборудование (например, реакторы, сушильные аппараты, мельницы и т.д.);
- данные о каждой серии (в т.ч. взвешивание, отмеривание и номера серий сырья, промежуточных продуктов или любых других материалов, использованных в производстве);
- фактические данные критических технологических параметров;
- данные о всех отобранных пробах;
- подписи непосредственных исполнителей и лиц, проверяющих или контролирующих каждую критическую операцию в данном процессе;
- результаты внутрипроизводственного и лабораторного контроля;
- фактический выход продукции на соответствующих стадиях или на определенный момент времени;
- данные об упаковке и маркировке для промежуточного продукта или АФС;
- образцы маркировки АФС или промежуточных продуктов, если они поставляются в готовом виде;
- любые отклонения, их оценку, результаты расследования (при необходимости) или ссылку на проведенное расследование (если оно хранится в виде отдельного протокола);
- результаты контроля при выпуске продукции.
6.53 Следует разработать инструкции по расследованию критических отклонений и несоответствия серии промежуточных материалов или АФС требованиям спецификаций. В это расследование следует включать и другие серии продукции, которые могут быть связаны с данным несоответствием или отклонением.
6.6 Протоколы лабораторного контроля
6.60 В протоколах лабораторного контроля должна быть полная информация о ходе всех испытаний, проведенных для подтверждения соответствия требованиям нормативной документации, в т.ч. данные наблюдений и анализов:
- описание полученных проб, в т.ч. наименование материала или место отбора пробы, номер серии или другой отличительный код, дата взятия образца, количество и дата поступления этой пробы в лабораторию;
- указание и/или ссылка на каждый использованный метод контроля;
- указание массы или размеров пробы для каждого испытания согласно принятой методике; данные или ссылка на приготовление и проведение испытания образцов сравнения, реактивов и стандартных растворов;
- необработанные в ходе каждого испытания данные в дополнение к графикам, таблицам и спектрам, полученным с использованием лабораторного оборудования с нумерацией (кодом), позволяющей определить испытуемое вещество и серию;
- вычисления с указанием единиц измерения, коэффициентов перевода и коэффициентов эквивалентности;
- результаты контроля и их сравнения с установленными критериями приемлемости;
- подпись ответственного лица, проводившего каждый вид контроля, и дату(ы) его проведения и
- дата и подпись второго лица, заверяющая точность, полноту и соответствие установленным требованиям.
6.61 Следует также составить протоколы с указанием:
- любых изменений в принятых методах контроля;
- периодической калибровки (поверки) лабораторных приборов, оборудования и записывающих устройств;
- испытаний на стабильность АФС;
- результатов исследований, не удовлетворяющих требованиям спецификации.
6.7 Рассмотрение протоколов серии продукции
6.70 Для определения соответствия промежуточного продукта или АФС установленным требованиям до выпуска или распространения данной серии продукции следует разработать инструкции по рассмотрению и утверждению (согласованию) протоколов на серию продукции и проведению лабораторного контроля качества продукции, в т.ч. упаковку и маркировку.
6.71 Протоколы на серию и лабораторные протоколы контроля критических технологических стадий до выпуска и реализации серии АФС должны быть рассмотрены и утверждены отделом(ами) контроля качества. Протоколы внутрипроизводственного и лабораторного контроля некритических технологических стадий могут быть рассмотрены квалифицированным производственным персоналом или другими отделами в соответствии с инструкциями, согласованными с отделом(ами) качества.
6.72 Все протоколы испытаний, не удовлетворяющих требованиям спецификации, отклонений от спецификаций и расследования этих случаев должны быть рассмотрены при выпуске серии в реализацию.
6.73 Служба (отделы) качества может передать ответственность и право выдачи разрешения на выпуск промежуточных продуктов производственному отделу (службе), за исключением случаев, когда промежуточные продукты поставляются за пределы зоны их контроля предприятия-производителя.
7 Работа с материалами
7.1 Общий контроль
7.10 Порядок получения, идентификации, карантинного хранения, обращения, отбора проб, проведения контроля и выдачи разрешения на использование или отклонение материалов должны быть определены в соответствующих инструкциях.
7.11 У производителей промежуточных продуктов и/или АФС должна быть система оценки поставщиков критических материалов.
7.12 Поставка материалов должна выполняться поставщиками, утвержденными отделом(ами) качества, в соответствии с согласованными спецификациями.
7.13 Если поставщик критического материала не является производителем данного материала, производитель промежуточных продуктов и/или АФС должен иметь информацию о наименовании и адресе этого производителя.
7.14 Замена поставщика критического сырья должна проводиться в соответствии с процедурой, изложенной в разделе 13 "Контроль изменений" настоящего приложения.
7.2 Приемка и карантин
7.20 Перед приемкой и при получении материалов на каждой упаковке или группе упаковок должна быть визуально проверена правильность маркировки (в т.ч. соответствие названия, используемого поставщиком и заказчиком, если они отличаются), сохранность упаковок, наличие поврежденных печатей, следов вскрытия и загрязнения упаковок. Материалы должны находиться на карантинном хранении до проведения отбора проб, их анализа (контроля) и получения разрешения на их использование.
7.21 До смешения поступивших материалов с имеющимися запасами (например, с растворителями или запасами, находящимися в хранилищах) следует установить их подлинность, при необходимости - провести контроль и получить разрешение на их использование. Для предотвращения неправильного размещения поступивших материалов среди уже хранящихся материалов должны быть разработаны специальные инструкции.
7.22 Следует исключить возможность перекрестного загрязнения нерасфасованной продукции, если она поставляется в емкостях, не предназначенных специально для нее. В качестве доказательства этого могут использоваться:
- паспорт очистки;
- контроль на наличие следов примесей;
- аудит поставщика.
7.23 Большие емкости для хранения и соответствующие коллекторы, линии по загрузке и выгрузке должны быть маркированы.
7.24 Каждая емкость или группа емкостей (серий) материалов должна иметь четкую маркировку (шифр, номер серии или поставки, контракта, товарной накладной). Эти номера должны использоваться при обозначении расположения каждой серии. Следует иметь систему обозначения статуса каждой серии.
7.3 Отбор проб и проведение испытаний поступивших материалов
7.30 Для подтверждения подлинности каждой серии материалов (за исключением материалов по пункту 7.32) следует провести хотя бы одно испытание. Вместо проведения последующих испытаний можно использовать аналитический паспорт поставщика при условии, что производитель имеет систему оценки поставщиков.
7.31 Система оценки поставщиков должна включать в себя доказательства (например, данные о качестве предыдущих поставок) того, что производитель может стабильно поставлять материалы, соответствующие требованиям спецификации. Прежде чем упростить процедуру собственных проверок, следует провести полный анализ не менее трех серий материала. Полный анализ должен проводиться через определенные интервалы времени и сравниваться с аналитическим паспортом. Достоверность аналитического паспорта необходимо регулярно проверять.
7.32 Не требуется проведение контроля технологических добавок, опасного или высокотоксичного сырья, других специальных материалов или материалов, передаваемых в другое подразделение под контролем заказчика, при наличии аналитического паспорта производителя, подтверждающего соответствие этого сырья установленным требованиям. Идентификация этих материалов выполняется визуальным осмотром упаковок, этикеток и регистрацией номеров серий. Если контроль таких материалов на месте не проводится, то это должно быть обосновано и оформлено документально.
7.33 Пробы должны быть репрезентативной выборкой серии материала, из которой они взяты. В методике отбора проб следует указывать число контейнеров, из которых должны быть взяты пробы, из какой части контейнера и в каком количестве они отбираются. Число контейнеров, из которых следует отобрать пробы, и количество проб определяются планом отбора проб. Следует учитывать критичность и стабильность свойств материала, информацию о качестве предыдущих поставок и количестве материала, необходимого для проведения анализа.
7.34 Отбор проб следует проводить в специально отведенном месте. Методика отбора проб должна предусматривать предотвращение загрязнения материала, из которого отбираются пробы, и других материалов.
7.35 Упаковки, из которых отбирают пробы, должны быть аккуратно вскрыты и закрыты. На упаковках должна быть нанесена маркировка, показывающая, что из этой упаковки были взяты пробы.
7.4 Хранение
7.40 При обращении с материалами и их хранении не допускаются ухудшение их качества, загрязнение и перекрестное загрязнение.
7.41 Материалы, хранящиеся в картонных барабанах, мешках или коробках, должны располагаться таким образом, чтобы было удобно проводить уборку и проверку. Не допускается складирование материалов на полу.
7.42 Период и условия хранения материалов не должны отрицательно влиять на их качество. Как правило, материалы, поступившие первыми, следует использовать в первую очередь.
7.43 Некоторые материалы можно хранить вне помещений при условии надлежащей упаковки, читаемой маркировки и выполнения необходимой очистки упаковок перед их вскрытием и использованием.
7.44 Отклоненные материалы должны иметь соответствующую маркировку и помещаться в карантин (изолятор брака) для исключения их несанкционированного использования в производстве.
7.5 Повторный контроль
7.50 При необходимости для определения пригодности материалов (например, после длительного хранения или в результате воздействия температуры или влажности) следует проводить повторный контроль.
8 Технологический и внутрипроизводственный контроль
8.1 Технологические операции
8.10 Условия взвешивания и отмеривания сырья для производства промежуточных продуктов и АФС не должны оказывать влияния на их пригодность. Устройства для взвешивания и отмеривания должны быть необходимой точности.
8.11 Если материал разделяется на части для последующего использования в производственных операциях, то он должен помещаться в специальную упаковку, маркировка которой содержит следующую информацию:
- наименование материала и/или код;
- номер, присвоенный при получении, или контрольный номер;
- массу или меру материала в новой упаковке и
- дату повторного контроля, при необходимости.
8.12 Выполнение критических операций по взвешиванию, отмериванию или разделению материала на порции должно быть заверено или проконтролировано эквивалентным образом. До начала работы производственный персонал должен убедиться в том, что все полученные материалы соответствуют данным, определенным в протоколе серии для производства данного промежуточного продукта или АФС.
8.13 Выполнение других критических действий должно быть заверено или проконтролировано эквивалентным образом.
8.14 На определенных стадиях технологического процесса необходимо сравнивать фактический выход продукции (материалов) с ожидаемым. Ожидаемый выход и допустимые отклонения должны быть определены на основании результатов лабораторных, опытных или производственных испытаний, проведенных предварительно. Отклонения от ожидаемого выхода, связанные с критическими технологическими стадиями, должны быть расследованы для определения их возможного влияния на качество серии продукции.
8.15 Все расхождения следует оформлять документально с указанием их причин. Любое критическое отклонение должно быть расследовано.
8.16 Назначение основных единиц оборудования должно быть указано на самом оборудовании или отражено в соответствующей документации, системе с компьютерным управлением и контролем или другим образом.
8.17 Следует организовать контроль материалов, предназначенных для повторного использования или переработки во избежание их несанкционированного использования.
8.2 Ограничения на время выполнения операций
8.20 Промышленным регламентом могут быть установлены ограничения на время выполнения операций (6.41). Отклонения от этих ограничений следует оформлять документально и проводить их оценку. Такие ограничения могут не потребоваться при проведении технологического процесса до достижения заданных значений параметров (например, доводка рН, гидрогенизация, сушка до значения, установленного спецификацией), так как в этом случае завершение реакций или стадий производства определяется при внутрипроизводственном отборе проб и контроле.
8.21 Условия хранения промежуточных продуктов должны обеспечивать их дальнейшую пригодность.
8.3 Внутрипроизводственный отбор проб и контроль
8.30 Для контроля и выполнения технологических стадий, которые влияют на показатели качества промежуточных продуктов и АФС, должны быть разработаны инструкции. Порядок проведения внутрипроизводственного контроля и критерии приемлемости определяются на основании информации, полученной при разработке технологического процесса или из предыдущего опыта производства.
8.31 Критерии приемлемости, вид и объем испытаний могут зависеть от вида производимых промежуточных продуктов или АФС, проводимой реакции или технологической стадии и степени влияния этого процесса на качество продукции. На ранних технологических стадиях допустим менее строгий внутрипроизводственный контроль; на более поздних технологических стадиях (например, на стадиях выделения и очистки) контроль может быть более жестким.
8.32 Порядок проведения внутрипроизводственного контроля на критических стадиях должен включать в себя контрольные точки и методы контроля, быть оформлен в письменном виде и согласован с отделом(ами) качества.
8.33 Внутрипроизводственный контроль может выполняться персоналом производственного подразделения, имеющим необходимую квалификацию, а наладка технологического процесса может выполняться без разрешения отдела(ов) качества, если она осуществляется в установленных допустимых пределах, согласованных с отделом(ами) качества. Результаты такого контроля в полном объеме должны быть документально оформлены и входить составляющей частью в протокол на серию продукции.
8.34 Порядок отбора проб промежуточных материалов, продуктов и АФС должен быть оформлен в виде письменных методик. Планы и методики отбора проб должны быть научно обоснованными.
8.35 Внутрипроизводственный отбор проб должен проводиться с использованием методик, предусматривающих предотвращение загрязнения материалов и других промежуточных продуктов или АФС. Применяемые методы должны обеспечивать целостность проб после их отбора.
8.36 Как правило, результаты внутрипроизводственного контроля, не соответствующие требованиям спецификаций, которые используются для проверки и/или наладки технологического процесса и не требуют проведения анализа причин отклонений.
8.4 Смешанные серии промежуточных продуктов или АФС
8.40 В данном Руководстве смешение определяется как процесс объединения материалов в рамках одной спецификации для производства однородного промежуточного продукта или АФС. Внутрипроизводственное смешение фракций одной серии (например, объединение нескольких загрузок центрифуги из одной кристаллизационной серии или объединение фракций из нескольких серий для дальнейшего производства) рассматривается как часть технологического процесса и не считается смешением.
8.41 Не допускается смешения серии, не удовлетворяющей требованиям спецификации, с другими сериями. Каждая серия, вносимая в смесь, должна быть произведена согласно установленным требованиям, индивидуально испытана, и ее соответствие спецификации должно быть подтверждено до смешения.
8.42 Как правило, смешиваются:
- малые серии для увеличения размера серии;
- остатки (т.е. относительно малые количества выделенного материала) из серий одного и того же промежуточного продукта или АФС для формирования одной серии.
При необходимости допускаются и другие случаи смешения материалов.
8.43 Процессы смешения следует контролировать и оформлять в документальном виде, а смешанная серия должна, при необходимости, испытываться на соответствие установленным требованиям.
8.44 В протоколе серии, полученной после смешения, должен прослеживаться процесс производства индивидуальных серий, формирующих эту смешанную серию.
8.45 Если физические характеристики АФС являются критическими (например, АФС предназначена для использования в твердых дозированных формах для принятия внутрь или в суспензиях), то операции по смешению должны быть аттестованы (валидированы) для подтверждения однородности окончательной серии. Аттестация (валидация) должна включать в себя испытание критических параметров (распределение частиц по размеру, объемная плотность при свободной засыпке и удельная плотность при уплотнении), на которые может повлиять процесс смешения.
8.46 Если смешение может оказать неблагоприятное воздействие на стабильность, то следует провести испытания стабильности окончательной серии.
8.47 Срок годности или дата проведения повторного контроля смешанной серии должны быть основаны на дате производства самой старой фракции или серии в смеси.
8.5 Контроль загрязнений
8.50 Допускается переносить остатки материалов в последующие серии того же промежуточного продукта или АФС при наличии соответствующего контроля. К ним относятся остатки, прилипающие к стенкам измельчителя; слой влажных кристаллов, остающийся в центрифуге после разгрузки, и неполное удаление жидкостей или кристаллов из технологической емкости после передачи материала на следующую технологическую стадию. Такой перенос не должен приводить к переносу продуктов распада или микробного загрязнения, которые могут оказать отрицательное влияние на чистоту АФС.
8.51 При выполнении технологических операций не следует допускать загрязнения промежуточных продуктов или АФС другими материалами.
8.52 После проведения очистки оборудования следует принять меры предосторожности, чтобы избежать загрязнения АФС.
9 Упаковка и маркировка АФС и промежуточных продуктов
9.1 Общие положения
9.10 Следует разработать и документально оформить методики на получение, идентификацию, карантинное хранение, отбор проб, проведение исследования и/или испытания и выдачу разрешения на использование, а также на обращение с упаковочными и печатными материалами.
9.11 Материалы для упаковки и маркировки должны соответствовать установленным требованиям. Материалы, не соответствующие этим требованиям, должны отклоняться для предотвращения их использования в операциях, для которых они непригодны.
9.12 Следует хранить протоколы на каждую поставку печатных и упаковочных материалов, содержащие информацию о поставке, осмотре или контроле этого материала, а также заключение о разрешении его использования или отклонения.
9.2 Упаковочные материалы
9.20 Упаковка должна обеспечивать надлежащую защиту промежуточного продукта или АФС от порчи или загрязнения, которые могут произойти во время перевозки или хранения.
9.21 Упаковки должны быть чистыми и, если это обусловлено видом промежуточного продукта или АФС, должны пройти обработку (дезинфекцию) для обеспечения пригодности их для дальнейшего использования. Упаковки не должны вступать в химические реакции и обладать аддитивными или абсорбирующими свойствами, чтобы не допустить изменения качества промежуточного продукта или АФС за установленные пределы.
9.22 При повторном использовании упаковки должны быть очищены в соответствии с утвержденными инструкциями, а все предыдущие этикетки удалены.
9.3 Выпуск и контроль печатных материалов
9.30 Доступ к местам хранения печатных материалов должен быть разрешен только лицам, имеющим соответствующие полномочия.
9.31 Порядок определения соответствия между количеством отпущенного, использованного и возвращенного печатного материала и оценки расхождений между количеством маркированных упаковок и отпущенных этикеток должен быть определен в инструкции. Факты несоответствия должны расследоваться, а результаты расследования должны утверждаться отделом(ами) контроля качества.
9.32 Весь лишний печатный материал, содержащий номер серии или другую печатную информацию о серии, должен быть уничтожен. Возвращенный печатный материал должен храниться таким образом, чтобы предотвратить его перепутывание и обеспечить надлежащую идентификацию.
9.33 Вышедший из употребления и устаревший печатный материал должен быть уничтожен.
9.34 Следует организовать контроль за работой устройств, предназначенных для печатания этикеток для упаковочных операций, чтобы гарантировать соответствие всех оттисков протоколу на серию продукции.
9.35 Следует организовать тщательный контроль этикеток, отпечатанных для серии, на их соответствие данной серии и установленным требованиям. Результат контроля должен быть оформлен документально.
9.36 Образец используемой отпечатанной этикетки должен быть включен в протокол серии продукции.
9.4 Операции по упаковке и маркировке
9.40 Следует разработать и оформить документально инструкции по использованию упаковочных и печатных материалов.
9.41 Следует разработать инструкции по выполнению маркировки, исключающие перепутывание печатных материалов. Операции по маркировке различных промежуточных продуктов или АФС должны быть разграничены физически или пространственно.
9.42 Этикетки, используемые на упаковках промежуточных продуктов или АФС, должны содержать наименование или идентификационный код, номер серии продукта и условия хранения (в том случае, когда эта информация является критической для обеспечения качества промежуточного продукта или АФС).
9.43 Если промежуточный продукт или АФС предназначаются для передачи за пределы зоны действия системы контроля их производителем, то маркировка также должна содержать название и адрес производителя, количество содержимого, специальные условия транспортирования и любые другие предусмотренные нормативной документацией требования. Если для промежуточных продуктов или АФС установлен срок годности, то он должен быть указан на маркировке и в аналитическом паспорте. Для промежуточных материалов или АФС, прошедших повторный контроль, дата повторного испытания должна быть указана на маркировке и в аналитическом паспорте.
9.44 Упаковочное и печатное оборудование должно быть проверено непосредственно перед использованием для гарантии того, что все материалы, не нужные для последующей упаковки, были удалены. Эта проверка должна быть отражена в протоколе серии продукции, журнале оборудования или другой документации.
9.45 В состав операций по упаковке входит проверка правильности маркировки первичных и вторичных упаковок промежуточных продуктов или АФС. Результаты этих проверок должны указываться в протоколах серии продукции или контроля качества продукции.
9.46 Промежуточные продукты или АФС, отправляемые за пределы зоны контроля производителя, должны быть опечатаны таким образом, чтобы при повреждении или отсутствии печати получатель обратил внимание на возможность замены содержимого.
10 Хранение и реализация
10.1 Хранение на складе
10.10 В помещениях для хранения материалов должны быть предусмотрены соответствующие условия (например, поддержание заданной температуры и влажности, при необходимости). Следует вести протоколы условий хранения, если они являются критическими для сохранения свойств материалов.
10.11 Во избежание случайного или несанкционированного использования находящихся в карантине, отклоненных, возвращенных или отозванных материалов должны быть выделены отдельные зоны для их временного хранения до принятия решения об их дальнейшем использовании (если другое не указано в документации).
10.2 Реализация
10.20 Реализация АФС и промежуточных продуктов третьим сторонам допускается только после получения разрешения отдела качества на их выпуск. В условиях карантина АФС и промежуточные продукты могут быть переведены в другое подразделение при наличии разрешения отдела контроля качества, соответствующей документации и организации надлежащего контроля.
10.21 Транспортирование АФС и промежуточных продуктов не должно оказывать влияния на их качество.
10.22 Особые условия транспортирования и хранения АФС и промежуточных продуктов должны быть указаны на этикетке.
10.23 Производитель должен убедиться в том, что подрядчик, осуществляющий перевозку АФС и промежуточных продуктов по контракту, знает и соблюдает требуемые условия транспортирования и хранения.
10.24 Для обеспечения возможности быстрого отзыва каждой серии продукции должна быть организована система отслеживания за ее реализацией.
11 Лабораторный контроль
11.1 Общий контроль
11.10 Служба качества должна иметь в своем распоряжении необходимые лабораторные помещения и оборудование.
11.11 Следует разработать и документально оформить методики по отбору проб, проведению испытаний, выдаче разрешения на использование или отклонение материалов, регистрации и хранению данных, полученных в лаборатории. Ведение протоколов и отчетов должно соответствовать подразделу 6.6.
11.12 Все спецификации, планы отбора проб и методики проведения испытаний должны быть научно обоснованными и гарантировать, что сырье, АФС и промежуточные продукты, а также печатные и упаковочные материалы соответствуют установленным стандартам качества и/или чистоты. Спецификации и методики испытаний должны соответствовать спецификациям и методикам, приведенным в регистрационном досье. Могут использоваться спецификации, не входящие в регистрационное досье. Спецификации, планы отбора проб и методики испытаний, в т.ч. изменения к ним, должны быть разработаны соответствующим подразделением, рассмотрены и согласованы отделом(ами) качества.
11.13 Спецификации для АФС должны быть разработаны в соответствии с действующими нормами и технологическим процессом, должны включать в себя контроль за примесями (например, органическими и неорганическими примесями и остаточным содержанием растворителей). Если АФС имеет спецификацию на микробиологическую чистоту, то должны быть установлены и соблюдены соответствующие пределы действия по общему числу микроорганизмов и нежелательным микроорганизмам. Если АФС имеет спецификацию на эндотоксины, то должны быть установлены и соблюдены соответствующие пределы действия по уровню эндотоксинов.
11.14 Контроль в лаборатории должен проводиться и оформляться в письменном виде в ходе его проведения. Любые отклонения от указанных методик должны регистрироваться и обосновываться.
11.15 Любые результаты, выходящие за рамки спецификаций, должны быть расследованы и оформлены в соответствии с принятой методикой, предусматривающей проведение анализа данных, оценку наличия существенной проблемы, определение путей решения этой проблемы и формулирование выводов из проведенного расследования. После получения результатов, выходящих за рамки спецификаций, повторные отборы проб и/или повторные испытания должны проводиться в соответствии с принятыми методиками.
11.16 Реактивы и стандартные растворы должны приготавливаться и маркироваться в соответствии с принятой методикой. Для аналитических реактивов и растворов следует применять маркировку "Использовать до".
11.17 Для производства АФС необходимо иметь первичные стандартные образцы и документацию о происхождении каждого стандартного образца. Данные о хранении и использовании каждого первичного стандартного образца согласно рекомендациям поставщика должны быть приведены в соответствующих протоколах. Первичные стандартные образцы, полученные из официально признанных источников и хранящиеся в соответствии с рекомендациями поставщика, используются без проведения испытаний.
11.18 При невозможности получения первичных стандартных образцов из официально признанных источников следует разработать внутренние стандартные образцы производителя. Для установления подлинности и чистоты первичного стандартного образца требуется выполнить соответствующий контроль. Проведение контроля должно быть оформлено документально.
11.19 Следует разработать порядок изготовления, установления, идентификации, испытаний, утверждения и хранения вторичных стандартных образцов. Пригодность каждой серии вторичного стандартного образца должна быть определена до момента первого использования сравнением его с первичным стандартным образцом. Каждая серия вторичного стандартного образца должна периодически проходить проверку (аттестацию) в соответствии с письменной методикой.
11.2 Проведение испытаний промежуточных продуктов и АФС
11.20 Для подтверждения соответствия каждой серии промежуточного продукта и АФС требованиям спецификаций должны быть проведены необходимые лабораторные испытания.
11.21 Для каждой АФС определяется состав примесей (идентифицированных и неидентифицированных), присутствующих в обычной серии, произведенной в ходе конкретного контролируемого технологического процесса. Описание примесей должно включать в себя их идентификацию или какие-либо качественные аналитические параметры, позволяющие ее идентифицировать (например, время удерживания), диапазон содержания каждой выявленной примеси и их классификацию (неорганическая, органическая, растворитель). Состав примесей зависит обычно от технологического процесса и природы АФС. Для АФС, получаемых из растительного сырья или из тканей животных, определение состава примесей обычно не требуется. Требования к АФС, получаемым биотехнологическим путем, содержатся в соответствующей нормативной документации.
11.22 Для выявления изменений в АФС, происходящих вследствие изменений в сырье, оборудовании, рабочих параметрах или технологическом процессе, состав примесей периодически следует сравнивать с составом примесей, указанным в регистрационном досье, или с данными, полученными ранее.
11.23 Следует проводить контроль микробной чистоты каждой серии промежуточного продукта и АФС в соответствии с установленными требованиями.
11.3 Аттестация (валидация) аналитических методик (см. раздел 12)
11.4 Аналитические паспорта
11.40 На каждую серию промежуточного продукта и/или АФС должен выдаваться подлинный аналитический паспорт.
11.41 Аналитический паспорт должен содержать информацию о наименовании промежуточного продукта или АФС, в т.ч. о его чистоте, номере серии и дате выпуска. Для промежуточных продуктов и АФС с установленным сроком годности последний должен быть указан на маркировке и в аналитическом паспорте. Для промежуточных продуктов или АФС с датой повторного контроля эта дата также должна быть указана на маркировке и/или в аналитическом паспорте.
11.42 В аналитическом паспорте должны быть перечислены все испытания, выполненные в соответствии с нормативными документами или требованиями заказчика, в т.ч. критерии приемлемости и полученные количественные результаты (при их наличии).
11.43 В аналитическом паспорте должна быть дата и подпись ответственного лица отдела контроля качества, а также название, адрес и телефон первичного производителя. Если анализ был проведен вторичным упаковщиком или производителем, аналитический паспорт должен содержать название, адрес, телефон вторичного упаковщика/производителя и ссылку на название первичного производителя.
11.44 Новые аналитические паспорта, выпускаемые вторичными упаковщиками/производителями, поставщиками или лицами, действующими от их имени, должны содержать название, адрес и телефон лаборатории, проводившей эти анализы. В них также должны быть указаны название, адрес первичного производителя и оригинал паспорта на эту серию продукции с приложением его копии.
11.5 Контроль стабильности АФС
11.50 Следует разработать и документально оформить программу непрерывного контроля стабильности АФС, результаты которого должны использоваться для подтверждения условий хранения, при пересмотре срока годности или срока проведения повторного контроля.
11.51 Методики испытаний на стабильность должны быть аттестованы.
11.52 Пробы для контроля стабильности следует хранить в упаковках, имитирующих коммерческие упаковки, например, для АФС, поставляемых в мешках, помещенных в картонные барабаны, пробы для контроля стабильности могут упаковываться в мешки из того же материала, помещенные в барабаны, меньшие по размеру и сделанные из похожего или такого же материала, как и для АФС, поступающих в реализацию.
11.53 Для подтверждения даты проведения повторного контроля или срока годности этой продукции первые три реализуемые серии продукции, как правило, должны быть испытаны на стабильность. Если результаты предыдущих испытаний показывают, что эта АФС сохранится стабильной еще не менее двух лет, разрешается использовать менее трех серий.
11.54 В программу контроля стабильности ежегодно должна включаться, по крайней мере, хотя бы одна серия производимой АФС (за исключением случаев, когда в этот год она не производилась).
11.55 Для АФС с коротким сроком годности испытания следует проводить чаще. Например, для биотехнологических/биологических и других АФС со сроком хранения один год и менее пробы на стабильность следует отбирать и испытывать ежемесячно в течение первых 3 мес и затем с интервалом в 3 мес. При наличии данных, подтверждающих сохранение стабильности АФС, может быть рассмотрен вопрос об увеличении периодичности испытаний, например, проведение испытаний через 9 мес).
11.56 Условия хранения при проведении испытаний на стабильность должны, по возможности, соответствовать требованиям нормативной документации к проведению этих испытаний.
11.6 Срок годности и дата повторного контроля
11.60 Если промежуточный продукт предназначен для перемещения за пределы зоны действия системы управления качеством производителя и на него установлен срок годности или дата повторного контроля, то следует иметь данные по стабильности этого материала (например, опубликованную информацию, результаты испытаний).
11.61 Срок годности АФС и дата повторного контроля должны быть основаны на анализе данных, полученных при испытаниях на стабильность. Обычно для АФС используется дата повторного контроля, а не дата срока годности.
11.62 Определение предварительного срока годности или даты повторного контроля АФС может основываться на данных опытных серий в следующих случаях:
1) при производстве опытных серий использовались методы, моделирующие технологический процесс при промышленном производстве;
2) качество АФС соответствует качеству продукции, которая будет производиться в промышленном масштабе.
11.63 Для проведения повторного контроля должен быть взят образец, репрезентативный для этой серии.
11.7 Архивные образцы
11.70 Упаковка и хранение архивных образцов предназначены для возможной оценки качества серий АФС в будущем, но не для проведения испытаний этих образцов на стабильность.
11.71 Маркированные архивные образцы из каждой серии АФС следует сохранять не менее одного года после истечения срока годности данной серии, определенного производителем, или в течение трех лет после даты реализации этой серии, в зависимости от того, какой срок больше. Для АФС с датой повторного контроля аналогичные архивные образцы следует хранить в течение трех лет после того, как эта серия была полностью реализована производителем.
11.72 Архивные образцы должны храниться в такой же или в эквивалентной упаковке, используемой для АФС, либо в упаковке, более защищенной, чем упаковка, в которой реализуется эта продукция. Количество архивных образцов должно быть достаточным для проведения не менее двух полных анализов в соответствии с требованиями Фармакопеи, либо, в случае отсутствия соответствующей фармакопейной статьи, двух полных анализов согласно спецификации.
12 Аттестация (валидация)
12.1 Политика проведения аттестации (валидации)
12.10 Общая политика предприятия, цели и подход к процессам аттестации, в т.ч. к аттестации технологических процессов, методов очистки, аналитических методов, методик проведения внутрипроизводственного контроля, систем с компьютерным управлением и контролем и лиц, ответственных за разработку, рассмотрение, согласование и оформление каждой стадии аттестации (валидации), должны быть оформлены в письменном виде.
12.11 Как правило, в ходе разработки процесса (или по имеющимся данным) должны быть определены критические параметры (характеристики) и пределы их изменений, необходимые для обеспечения надежной работы. К таким параметрам относятся:
- критические характеристики, которые должны быть указаны для данного продукта (АФС);
- технологические параметры, которые могут повлиять на критические характеристики качества АФС;
- пределы изменения каждого критического параметра процесса, которые будут использоваться в ходе текущего производственного контроля.
12.12 Операции, которые считаются критическими для качества и чистоты АФС, подлежат аттестации.
12.2 Документация по аттестации (валидации)
12.20 Для каждого процесса, подлежащего аттестации, следует разработать методику проведения аттестации, согласованную службой качества и другими отделами, имеющими к ней отношение.
12.21 Методика аттестации определяет критические операции (стадии) технологического процесса и критерии приемлемости, а также виды аттестации (например, ретроспективная, перспективная, текущая) и количество производственных циклов, необходимых для проведения аттестации.
12.22 Отчет о проведении аттестации должен содержать ссылку на методику аттестации (с указанием всех замеченных отклонений) и выводы с рекомендациями по устранению недостатков.
12.23 Любые отклонения от методики аттестации должны быть оформлены в письменном виде и утверждены.
12.3 Этапы аттестации (валидации)
12.30 До проведения работ по аттестации процессов следует выполнить аттестацию критического оборудования и вспомогательных систем в необходимом объеме. Как правило, такая аттестация проводится в несколько этапов:
- аттестация проекта (DQ; design qualification) - документальное подтверждение того, что проект производства (в т.ч. помещения, оборудование или вспомогательные системы) соответствует заданию на проектирование и действующим нормам;
- аттестация установленного оборудования (IQ; Installation Qualification) - документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы соответствуют утвержденному проекту, заданию на проектирование и действующим нормам;
- аттестация функционирующего оборудования (OQ; Operational Qualification) - документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы функционируют надлежащим образом в требуемом режиме;
- аттестация в эксплуатации (PQ; Performance Qualification) - документальное подтверждение того, что оборудование, помещения и вспомогательные системы могут работать совместно с нужной эффективностью и воспроизводимостью в соответствии с утвержденными требованиями и характеристиками технологического процесса.
12.4 Аттестация (валидация) процесса
12.40 Аттестация (валидация) процесса - документальное подтверждение того, что процесс, протекающий в пределах установленных параметров, обеспечивает эффективное и воспроизводимое производство промежуточных продуктов и АФС, удовлетворяющих установленным требованиям и показателям качества.
12.41 Из трех видов аттестации (валидации) предпочтительным является перспективная аттестация (валидация).
12.42 Перспективная аттестация (валидация) применяется для всех процессов производства АФС, определенных в пункте 12.12. Перспективная аттестация (валидация), используемая для процесса производства АФС, должна быть завершена до выпуска на рынок готового лекарственного средства, произведенного из этого АФС.
12.43 Текущая аттестация (валидация) может проводиться при отсутствии данных о повторных технологических циклах. Это может быть обусловлено производством только ограниченного числа серий АФС, нерегулярным производством серий АФС или производством АФС с использованием аттестованного (валидированного) процесса, в который были внесены изменения. До завершения текущей аттестации (валидации) серии АФС могут быть выпущены и использованы для производства готовых лекарственных средств для их реализации на рынке на основании тщательного непрерывного контроля и проведения испытаний этих серий АФС.
12.44 Ретроспективная аттестация (валидация) применяется как исключение для хорошо отлаженных процессов, которые не претерпевали значительных изменений для качества АФС, из-за изменений в сырье, оборудовании, системах, устройствах или в данном технологическом процессе. Этот вид аттестации (валидации) может использоваться, если:
1) определены критические характеристики качества и критические параметры процесса;
2) установлены соответствующие внутрипроизводственные критерии приемлемости и контроля;
3) отсутствовали существенные сбои в ходе процесса (качестве продукции) по причинам, не связанным с ошибками оператора или отказами оборудования;
4) установлен состав примесей для существующих АФС.
12.45 Серии продукции, отобранные для проведения ретроспективной аттестации (валидации), должны являться репрезентативной выборкой всех серий, произведенных за рассматриваемый период, в т.ч. всех серий, которые не удовлетворяли требованиям спецификаций. Число этих серий должно быть достаточным для подтверждения стабильности процесса. При проведении ретроспективной аттестации (валидации) можно использовать архивные образцы.
12.5 Программа аттестации (валидации) процесса
12.50 Число производственных циклов для проведения аттестации (валидации) зависит от сложности процесса или характера изменения параметров процесса. Для проведения перспективной и текущей аттестации (валидации) следует использовать не менее трех последовательно произведенных серий продукции надлежащего качества. Однако могут быть случаи, при которых для доказательства стабильности процесса могут потребоваться дополнительные производственные циклы (например, сложные или длительные процессы производства АФС). При ретроспективной валидации для подтверждения стабильности процесса исследуют данные 10-30 последовательных серий продукции. При наличии обоснования это число может быть уменьшено.
12.51 В ходе аттестации (валидации) процесса следует контролировать его критические параметры. Параметры процесса, не связанные с качеством продукции (например, расход энергии или срок использования оборудования), можно не включать в процесс аттестации (валидации).
12.52 Аттестация (валидация) процесса должна подтвердить, что установленный состав примесей АФС находится в допустимых пределах. Состав примесей (качественный и количественный) должен быть сравним или лучше состава примесей, установленного по предыдущим данным; или, где применимо, быть лучше состава примесей, установленного при разработке технологического процесса; или профиля примесей в сериях продукции, предназначенных для проведения клинических или токсикологических исследований.
12.6 Периодическая оценка аттестованных (валидированных) систем и процессов
12.60 Системы и процессы следует периодически оценивать для подтверждения того, что они продолжают работать в аттестованном (валидированном) режиме. Повторная аттестация (валидация) не требуется, если в систему или процесс не было внесено никаких существенных изменений и анализ качества продукции подтверждает, что эта система или процесс стабильно производят продукцию в соответствии с установленными требованиями.
12.7 Аттестация (валидация) методов очистки
12.70 Как правило, следует проводить аттестацию (валидацию) методов очистки. Обычно аттестация (валидация) методов очистки проводится для случаев, когда загрязнение или перенос материалов представляют наибольший риск для качества АФС. Например, на ранних стадиях производства может оказаться ненужной аттестация (валидация) методов очистки оборудования, если все остатки удаляются последующими операциями очистки.
12.71 Аттестация (валидация) методов очистки должна отражать фактический характер использования оборудования. Если на одном и том же оборудовании производятся различные АФС или промежуточные продукты, и очистка этого оборудования проводится одним и тем же способом, то для аттестации (валидации) очистки может быть выбран репрезентативный образец промежуточного продукта или АФС. Этот выбор должен быть основан на свойствах растворимости вещества и трудности для его очистки, а также на расчете допустимых пределов остатков на основе его активности, токсичности и стабильности.
12.72 Методика аттестации (валидации) очистки должна содержать характеристику оборудования, подлежащего очистке, порядок очистки, перечень материалов, допустимые пределы чистоты, параметры, которые следует контролировать, и применяемые аналитические методы. В методике также следует указать вид отбираемых проб, методы их отбора и маркировки.
12.73 Для определения растворимых и нерастворимых остатков при отборе проб следует использовать такие методы, как взятие мазков, смывов, или альтернативные методы (например, прямую экстракцию). Используемые методы отбора проб должны позволять проводить количественное определение остатков на поверхности оборудования после его очистки. Отбор проб взятием мазков может быть не всегда применим, если поверхности контакта с продуктом являются труднодоступными из-за особенностей конструкции оборудования и/или ограничений процесса (например, внутренние поверхности шлангов, труб, сосудов, реакторов с маленькими отверстиями) или при работе с токсическими материалами, а также с малогабаритным сложным оборудованием (например, с измельчителями и псевдоожижителями).
12.74 Следует использовать аттестованные (валидированные) аналитические методы, обладающие достаточной чувствительностью для определения допустимого уровня остатков или загрязнений. Для каждого метода следует определить его воспроизводимость. Предельно допустимые количества остатков должны быть реальными, достижимыми, обоснованными и основываться на остатке наиболее опасного вещества. Предельно допустимые уровни остатков могут устанавливаться на основе минимальной известной фармакологической, токсикологической или физиологической активности АФС или ее наиболее опасного компонента.
12.75 Для процессов, в которых существует ограничение на общее микробное загрязнение или уровень эндотоксинов в АФС, или других процессов, в которых такое загрязнение может представлять опасность (например при производстве нестерильных АФС, используемых в производстве стерильных препаратов), аттестация (валидация) очистки (дезинфекции) оборудования должна включать в себя определение уровня загрязнения по микроорганизмам и эндотоксинам.
12.76 После проведения аттестации (валидации) методы очистки должны контролироваться с определенней периодичностью для подтверждения того, что они сохраняют эффективность при использовании в текущем производстве. Чистоту оборудования можно контролировать аналитическими методами, а там, где это допускается, - с помощью визуального осмотра. Визуальный осмотр позволяет определять большие скопления загрязнений на малых площадях, которые могут оставаться необнаруженными при отборе проб и/или проведении анализов
12.8 Аттестация (валидация) аналитических методов
12.80 Следует предусматривать аттестацию (валидацию) аналитических методов, за исключением фармакопейных или других утвержденных методов. В каждом конкретном случае следует подтвердить и оформить документально пригодность каждого применяемого аналитического метода.
12.81 При аттестации (валидации) методов следует проверять характеристики, включенные в Руководство ICH по валидации аналитических методов. Объем исследований, выполняемых при аттестации (валидации) аналитических методов, должен соответствовать целям анализа и стадии процесса производства АФС.
12.82 Перед началом аттестации (валидации) аналитических методов следует провести аттестацию (валидацию) соответствующего аналитического оборудования.
12.83 Протоколы всех изменений, вносимых в аттестованный (валидированный) аналитический метод, следует сохранять. В протоколах должны быть указаны причина, из-за которой было сделано изменение, и данные, подтверждающие, что точность и достоверность результатов, полученных после внесения этого изменения, соответствуют характеристикам аттестованного метода.
13 Контроль изменений
13.10 На предприятии должна быть введена и документально оформлена система контроля изменений, способных повлиять на производство и качество промежуточных продуктов и АФС.
13.11 Документальное оформление, рассмотрение и утверждение изменений в сырье, спецификациях, аналитических методах, помещениях, вспомогательных системах, оборудовании (в т.ч. в компьютерном оборудовании), технологических стадиях, печатных и упаковочных материалах и компьютерных программах должны выполняться по утвержденным инструкциям.
13.12 Все предложения по изменениям, касающимся соблюдения требований настоящего стандарта, должны быть подготовлены, рассмотрены и согласованы соответствующими подразделениями и отделом(ами) службы качества.
13.13 Следует оценивать возможное влияние предложенных изменений на качество промежуточных продуктов и АФС. При этом может проводиться классификация изменений для определения объема работ при аттестации. Изменения могут быть классифицированы (например, как незначительные и существенные) в зависимости от их характера и степени влияния, которые эти изменения могут оказать на данный процесс. Следует определить и обосновать, какие дополнительные испытания и аттестационные (валидационные) исследования необходимы для подтверждения возможности изменений в аттестованном (валидированном) процессе.
13.14 При внесении изменений следует принять меры по пересмотру всех документов, на содержание которых влияют эти изменения.
13.15 После внесения изменений следует оценить качество первых серий продукции, произведенных или испытанных с учетом этих изменений.
13.16 Следует оценить возможность влияния критических изменений на установленные сроки годности или дату повторного контроля. При необходимости образцы промежуточных продуктов или АФС, произведенных по модифицированному процессу, могут быть включены в программу ускоренного определения стабильности и/или программу непрерывного контроля стабильности.
13.17 Все производители готовых лекарственных средств, которым поставляются АФС, должны быть извещены об изменениях, прошедших в установленных методах производства и контроля качества, способных оказать влияние на качество АФС.
14 Отклонение и переработка материалов
14.1 Отклонение
14.10 Следует выполнить маркировку промежуточных продуктов и АФС, не удовлетворяющих требованиям спецификаций, и поместить их на карантинное хранение. Эти промежуточные продукты и АФС могут быть вновь направлены в производство или на переработку в приведенном ниже порядке. Окончательное размещение об использовании (утилизации) отклоненных материалов должно быть оформлено документально.
14.2 Повторная обработка
14.20 Как правило, допускается повторное направление промежуточных продуктов и АФС (в т.ч. материалов, не удовлетворяющих требованиям спецификации) в производство и их повторная обработка путем повторения стадии кристаллизации или других физических или химических стадий обработки (например, дистилляция, фильтрация, хроматография, измельчение), входящих в технологический процесс. Однако в том случае, если такие операции выполняются при производстве большинства серий, они должны быть включены в производство в качестве отдельной стадии принятого технологического процесса.
14.21 Продолжение технологической стадии после того, как внутрипроизводственный контроль показал, что эта стадия еще не завершена, рассматривается как часть процесса и не считается повторной обработкой.
14.22 Введение непрореагировавшего материала снова в процесс и повторение химической реакции считаются повторной обработкой, если такая операция не является частью технологического процесса. Такой повторной обработке должна предшествовать тщательная оценка, гарантирующая, что качество промежуточных продуктов и АФС не ухудшится вследствие возможного образования побочных продуктов и веществ при дальнейшем протекании реакции.
14.3 Переработка
14.30 Перед принятием решения о переработке серии, не соответствующей установленным требованиям, следует провести расследование причин такого несоответствия.
14.31 Для серий продукции, полученных в результате переработки, следует провести ее оценку, испытания и, при необходимости, - испытания на стабильность с документальным оформлением для подтверждения соответствия ее качества качеству продукции, произведенной по базовой технологии. Для методик переработки может использоваться подход текущей аттестации (валидации). Это позволяет отработать методику и определить ожидаемые результаты. Допускается единичный выпуск серии переработанной продукции на основании отчета о ее производстве.
14.32 Следует разработать методики сравнения состава примесей в каждой серии переработанной продукции с составом примесей в сериях продукции, произведенных по базовой технологии. Если для описания характеристик серии переработанной продукции недостаточно обычного набора аналитических методов, следует применять дополнительные методы.
14.4 Регенерация материалов и растворителей
14.40 Допускается регенерация реактивов, промежуточных продуктов или АФС (например, из маточных растворов или фильтратов) при наличии утвержденных методик регенерации и соответствия качества регенерированных материалов установленным требованиям.
14.41 Допускается регенерация и повторное использование растворителей в том или другом процессе при условии контроля процессов их регенерации и подтверждения соответствия растворителей установленным требованиям до их повторного использования или смешения с другими материалами.
14.42 Допускается смешение свежеприготовленных и регенерированных растворителей и реактивов, если при испытаниях была показана их пригодность во всех технологических процессах, в которых они могут использоваться.
14.43 Использование регенерированных растворителей, маточных растворов и других регенерированных материалов должно оформляться документально.
14.5 Возврат
14.50 Возвращенные промежуточные продукты или АФС должны иметь соответствующую маркировку и помещаться на карантинное хранение.
14.51 Если условия, в которых хранились или транспортировались возвращенные промежуточные продукты или АФС до или в ходе их возврата, или состояние их упаковок вызывают сомнения относительно качества этих материалов, возвращенные промежуточные продукты или АФС должны быть переработаны, повторно обработаны или уничтожены в установленном порядке.
14.52 Следует хранить протоколы о всех возвращенных промежуточных продуктах или АФС. Протокол каждого возврата должен содержать:
- наименование и адрес грузополучателя;
- наименование промежуточного продукта или АФС, номер серии и возвращенное количество;
- причину возврата;
- указание на использование или уничтожение возвращенного промежуточного продукта или АФС.
15 Рекламации и отзывы
15.10 Все рекламации по качеству, полученные в устной или письменной форме, должны регистрироваться и расследоваться согласно письменной инструкции.
15.11 Протоколы рассмотрения рекламаций должны включать в себя:
- имя и адрес лица, предъявившего рекламацию;
- имя, должность и телефон лица, принявшего эту рекламацию;
- суть рекламации (с указанием наименования и номера серии АФС);
- дату поступления рекламации;
- первоначально принятые меры (с указанием ответственных лиц и даты);
- дальнейшие действия;
- ответ на рекламацию (с датой отправки ответа);
- окончательное решение по серии или партии промежуточного материала или АФС.
15.12 Для оценки тенденций, частоты и серьезности рекламаций по данному продукту с точки зрения принятия дополнительных и немедленных мер по их исправлению протоколы рекламаций должны сохраняться.
15.13 Следует разработать и оформить в письменном виде инструкции, определяющие условия, при которых следует принимать решение об отзыве промежуточного продукта или АФС.
15.14 В инструкции по отзыву продукции должны быть определены лица, участвующие в анализе поступившей информации; процедуры начала отзыва продукции; круг лиц, которых следует проинформировать об отзыве, и порядок использования (утилизации) отозванного материала.
15.15 В случае серьезной или потенциальной опасности для жизни населения следует проинформировать местные, национальные и/или международные компетентные органы и обратиться к ним с просьбой о содействии.
16 Работа по контракту (в т.ч. проведение анализов)
16.10 Все производители, работающие по контракту (в т.ч. лаборатории), должны выполнять требования настоящего стандарта. Особое внимание должно быть уделено вопросам предотвращения перекрестного загрязнения и обеспечению прослеживаемости.
16.11 Заказчик должен оценить способность подрядчика осуществлять на месте делегированные ему операции в соответствии с требованиями настоящего стандарта.
16.12 Ответственность заказчика и подрядчика за соблюдение требований настоящего стандарта, в т.ч. за меры по обеспечению качества, должна быть определена в контракте (договоре) между ними.
16.13 Договор должен позволять заказчику проводить аудит производства подрядчика на его соответствие настоящему стандарту.
16.14 Если разрешен субподряд, то подрядчик не должен передавать третьей стороне никакой части работы, порученной ему заказчиком, без согласования с ним.
16.15 Производственные и лабораторные протоколы должны храниться в местах осуществления этого вида деятельности и быть легко доступными.
16.16 Не допускается внесение изменений в технологический процесс, оборудование, спецификации, методы испытаний и др., относящиеся к предмету договора, без согласования с заказчиком.
17 Реализация, хранение, переупаковка и перемаркировка
17.1 Область применения
17.10 Этот раздел относится к любым действиям (за исключением производства), связанным с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС.
17.11 Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны соблюдать требования настоящего стандарта и, в частности, данного Руководства.
17.2 Прослеживаемость реализуемых промежуточных продуктов или АФС
17.20 При реализации, хранении, переупаковке и перемаркировке необходимо обеспечивать полную прослеживаемость реализуемых промежуточных продуктов или АФС. Для этого следует иметь и хранить следующие документы:
- реквизиты исходного производителя;
- адрес исходного производителя;
- заказы (договоры) на поставку;
- накладные (транспортные документы);
- документы о получении грузов;
- наименование или обозначение промежуточных продуктов или АФС;
- номер серии производителя;
- документацию на транспортирование и реализацию;
- все подлинные аналитические паспорта, в т.ч. паспорта исходного производителя;
- дату повторного контроля или срок годности.
17.3 Управление качеством
17.30 Реализация, хранение, переупаковка и перемаркировка промежуточных продуктов или АФС должны выполняться в соответствии с документально оформленной системой управления качеством (подраздел 3.21 настоящего стандарта).
17.4 Переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС
17.40 Во избежание перепутывания и потери качества и чистоты промежуточных продуктов или АФС переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС и их контроль должны выполняться согласно требованиям данного Руководства.
17.41 Во избежание прямого и перекрестного загрязнения их переупаковка должна проводиться при требуемых условиях окружающей среды.
17.5 Стабильность
17.50 Для подтверждения установленных сроков годности или даты повторного контроля исследования по стабильности должны проводиться в случаях, если АФС или промежуточный продукт переупаковываются в упаковку другого типа, отличающуюся от используемой производителем.
17.6 Передача информации
17.60 При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует передавать заказчикам всю информацию, получаемую от производителя АФС или промежуточных продуктов, касающуюся качества продукции и решений уполномоченных органов, а также передавать производителям информацию от заказчиков.
17.61 При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует предоставлять потребителю наименование исходного производителя АФС или промежуточных продуктов и номера поставляемых серий продукции.
17.62 По требованию уполномоченного органа должна предоставляться информация об исходном производителе промежуточных продуктов или АФС. Исходный производитель может иметь контакт с уполномоченным органом непосредственно или через своих представителей.
17.63 Следует соблюдать специальные требования к аналитическим паспортам, приведенные в подразделе 11.4.
17.7 Работа с рекламациями и отзывами
17.70 Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны хранить протоколы всех поступивших рекламаций и отзывов в соответствии с разделом 15 настоящего Руководства.
17.71 При необходимости эти лица и организации вместе с производителем АФС или промежуточного продукта должны рассмотреть рекламацию для принятия решения о дальнейших действиях, в которых могут участвовать другие потребители (заказчики), получавшие этот АФС или промежуточный продукт, уполномоченный орган или все заинтересованные стороны. Соответствующей стороной должно быть проведено расследование случая рекламации или отзыва, результаты которого оформляются документально.
17.72 Если рекламация касается производителя АФС или промежуточного продукта, протокол расследования должен содержать любые данные, полученные от этого производителя АФС или промежуточного продукта (в т.ч. дату и необходимую информацию).
17.8 Работа с возвратами
17.80 Работа с возвратом продукции должна проводиться в соответствии с пунктом 14.52 настоящего Руководства. Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны вести документацию по возвращенным АФС и промежуточным продуктам.
18 АФС, производимые путем культивирования клеток (ферментации)
18.1 Общие положения
18.10 В настоящем разделе приведены специальные правила производства и контроля качества АФС или промежуточных продуктов путем культивирования (ферментации) клеток с использованием природных или рекомбинантных организмов, которые не были в должной мере отражены в предыдущих разделах. Этот раздел нельзя отделить от общего содержания данного документа. Все требования, упомянутые в настоящем стандарте, остаются в силе. Следует обратить внимание на то, что принципы ферментации для "классических" процессов получения низкомолекулярных соединений и процессов, использующих рекомбинантные и нерекомбинантные организмы для получения белков и/или полипептидов, одинаковы, хотя при этом уровень контроля этих процессов будет различным. Там, где это имеет значение, в данном разделе указывается на эти различия. В целом уровень контроля биотехнологических процессов, используемых для производства белков и полипептидов, должен быть более строгим, чем контроль над классическими процессами ферментации.
18.11 Термин "биотехнологический процесс" относится к использованию клеток или организмов, которые для производства АФС были получены или модифицированы с использованием рекомбинантной ДНК, гибридомной или какой-либо другой технологии. АФС, произведенные биотехнологическими методами, обычно представляют собой высокомолекулярные соединения, такие как белки и полипептиды, для которых в этом разделе приведены специфические требования. С использованием технологии рекомбинантной ДНК могут быть также произведены некоторые АФС с низкими молекулярными массами, например, антибиотики, аминокислоты, витамины и углеводы. Уровень контроля за такими АФС соответствует правилам, применяемым для классической ферментации.
18.12 Термин "классическая ферментация" относится к процессам, использующим для производства АФС микроорганизмы, существующие в природе или модифицированные традиционными методами (радиацией, химическим мутагенезом). АФС, получаемые методом "классической ферментации", обычно являются низкомолекулярными соединениями, такими как антибиотики, аминокислоты, витамины и углеводы.
18.13 Получение АФС и промежуточных продуктов из культуры клеток или методом ферментации включает в себя такие биологические процессы, как культивирование клеток или экстракция и очистка веществ из живых организмов. Следует обратить внимание на то, что могут существовать и дополнительные стадии (например, физико-химическая модификация), которые являются частью технологического процесса. Используемые исходные материалы (питательные среды, компоненты буфера) могут обеспечивать возможность роста микробных загрязнений. В зависимости от природы, метода приготовления и целей использования АФС или промежуточного продукта на определенных стадиях производства может потребоваться контроль микробного, вирусного загрязнения и/или уровня эндотоксинов в такой продукции.
18.14 Для обеспечения качества АФС и промежуточных продуктов на всех стадиях производства должен быть установлен надлежащий контроль. Так как это Руководство начинается со стадии культивирования клеток (ферментации), предыдущие стадии (например, поддержание банка клеток) должны проводиться при наличии надлежащего внутрипроизводственного контроля. Настоящее Руководство охватывает процесс культивирования (ферментации) клеток с момента, когда из банка клеток поступает емкость с культурой клеток для использования в производстве.
18.15 Для сведения риска загрязнения к минимуму следует использовать необходимое оборудование и проводить контроль окружающей среды. Критерии приемлемости качества и периодичность проведения контроля окружающей среды зависят от технологической стадии и условий производства (открытая, закрытая или замкнутая система).
18.16 В целом технологический контроль должен учитывать:
- поддержание в рабочем состоянии рабочего банка клеток;
- правильную инокуляцию и развитие культуры клеток;
- контроль критических рабочих параметров во время ферментации/ культивирования клеток;
- непрерывный контроль процесса роста клеток, их жизнеспособности (для большинства процессов культивирования клеток) и продуктивности;
- методики сбора и очистки, при которых происходит удаление клеток, клеточного дебриса и компонентов питательной среды с одновременной защитой промежуточного продукта или АФС от загрязнения (в частности, микробиологического характера) и потери качества;
- непрерывный контроль микробного загрязнения и, при необходимости, уровня эндотоксинов на определенных стадиях производства, и
- вопросы защиты от вирусного загрязнения, описанные в соответствующей нормативной документации (Руководстве ICH Q5A "Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения" (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin)).
18.17 Должно быть показано удаление остатков среды, белков клетки-хозяина, других примесей и загрязнений, связанных с технологическим процессом.
18.2 Поддержание банков клеток и ведение протоколов
18.20 Доступ к банкам клеток должен быть ограничен лицами, имеющими на это полномочия.
18.21 Банки клеток должны храниться в специально разработанных условиях, обеспечивающих поддержание жизнеспособности и предотвращающих загрязнение клеток.
18.22 Следует вести и сохранять протоколы использования образцов из банков клеток и условия их хранения.
18.23 Банки клеток должны периодически проходить контроль пригодности их использования.
18.24 Более подробная информация по содержанию банков клеток изложена в соответствующей нормативной документации (Руководство ICH Q5A "Качество биотехнологических продуктов. Выделение и характеристика клеточных субстратов, используемых для производства биотехнологических/биологических продуктов" (Quality of Biotechnological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products).
18.3 Культивирование клеток (ферментация)
18.30 Закрытые системы следует применять, по возможности, в тех случаях, когда необходимо добавлять клеточные субстраты, среды, буфер и газы в асептических условиях. Если первичная инокуляция, последующий перенос или добавки (среды, буфер) производятся в открытых сосудах, для снижения риска заражения должны быть разработаны и применяться соответствующие методики и контроль.
18.31 Если микробное загрязнение может оказать влияние на качество АФС, манипуляции с использованием открытых сосудов следует проводить в стерильных боксах или других устройствах, обеспечивающих контроль условий окружающей среды.
18.32 При работе с культурами клеток персонал должен быть одет в специальную одежду и соблюдать специальные меры предосторожности.
18.33 Следует проводить непрерывный контроль критических рабочих параметров (например, температуры, рН, скорости перемешивания, добавления газов, давления), который позволяет гарантировать соответствие условий установленным параметрам процесса. Следует также постоянно контролировать рост клеток, жизнеспособность (для большинства культивируемых клеток) и их продуктивность. В зависимости от типа процесса критические параметры могут изменяться, а в случае классической ферментации некоторые параметры можно не контролировать (например, жизнеспособность клеток).
18.34 Оборудование, используемое для культивирования клеток, после окончания процесса должно быть очищено и стерилизовано. Оборудование для проведения ферментации также должно быть очищено, дезинфицировано или стерилизовано.
18.35 Для предотвращения неблагоприятного влияния на качество АФС следует стерилизовать среды культивирования перед использованием.
18.36 Методики обнаружения загрязнения и принятия соответствующих мер должны быть оформлены документально. К ним относятся методики определения влияния загрязнения на продукцию, методы очистки оборудования и приведение оборудования в состояние готовности для дальнейшего использования. Посторонние микроорганизмы, обнаруженные в ходе процесса ферментации, должны быть идентифицированы, а влияние их на качество продукции должно быть оценено. При решении вопроса о судьбе произведенной продукции результаты таких оценок должны приниматься во внимание.
18.37 Протоколы о случаях загрязнения следует хранить.
18.38 После очистки универсального оборудования между циклами производства различной продукции может потребоваться проведение дополнительных испытаний с целью снижения риска перекрестных загрязнений.
18.4 Сбор, выделение и очистка
18.40 Операции по сбору материала (удаление клеток или клеточных компонентов или сбор клеточных компонентов после разрушения клеток) должны проводиться на оборудовании и в зонах, проект (конструкция) которых должен предусматривать снижение риска загрязнения до минимума.
18.41 Следует разработать методики сбора и очистки, приводящие к удалению или инактивации организмов-продуцентов, клеточного дебриса и компонентов среды (оказывающих при этом минимальное влияние на деградацию, загрязнение и потерю качества) и гарантирующие выход промежуточного продукта и АФС надлежащего качества.
18.42 После использования оборудование должно быть очищено и дезинфицировано в установленном порядке. Производство нескольких последовательных серий промежуточного продукта и АФС без промежуточной очистки оборудования допускается только в том случае, если это не оказывает влияния на их качество.
18.43 При использовании открытых систем очистка должна проводиться в условиях, обеспечивающих сохранение качества продукции.
18.44 Если оборудование используется для производства нескольких видов продукции, могут применяться дополнительные виды контроля, такие как использование специальных хроматографических смол или проведение дополнительных испытаний.
18.5 Удаление вирусов (стадии инактивации)
18.50 Более полная информация дана в Руководстве ICH Q5A "Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения" (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
18.51 Стадии удаления и инактивации вирусов являются критическими для некоторых технологических процессов и должны проводиться в рамках параметров, установленных при их аттестации (валидации).
18.52 Следует предусматривать меры предосторожности для предотвращения потенциального заражения вирусами, начиная со стадий до и после удаления (инактивации) вирусов. Открытая обработка должна выполняться в зонах, отделенных от других технологических операций и имеющих отдельную систему вентиляции.
18.53 Как правило, для различных стадий очистки не используется одно и то же оборудование. Если оборудование используется для разных стадий очистки, перед повторным использованием оно должно быть очищено и дезинфицировано соответствующим образом. Следует предпринимать меры, предотвращающие перенос вирусов с предыдущих стадий (например, через оборудование или окружающую среду).
19 АФС, предназначенные для проведения клинических исследований
19.1 Общие положения
19.10 При производстве новых АФС, используемых для проведения клинических исследований при их разработке, могут применяться не все виды контроля, рассмотренные в предыдущих разделах данного Руководства. В настоящем разделе приведены специальные требования к этим АФС.
19.11 Контроль при производстве АФС, предназначенных для клинических исследований, должен соответствовать той стадии разработки лекарственного средства, в состав которого она входит. Методы производства и контроля качества таких АФС должны быть достаточно гибкими для того, чтобы предусмотреть изменения, вносимые в них по мере получения новых знаний о технологическом процессе и продвижении лекарственного средства с доклинических стадий к клиническим стадиям исследований. Если разработка лекарственного препарата достигает стадии, на которой эта АФС производится для применения в лекарственном препарате, предназначенном для проведения клинических исследований, производители должны гарантировать, что производственные мощности, соответствующие методы производства и контроля позволяют гарантировать качество этой АФС.
19.2 Качество
19.20 К производству АФС, предназначенных для использования при проведении клинических исследований, должны применяться требования настоящего стандарта с надлежащей процедурой выдачи разрешения на реализацию каждой серии.
19.21 Для выдачи разрешения на реализацию или отклонение каждой серии АФС, предназначенной для использования в клинических исследованиях, должен(ы) быть образован(ы) отдел(ы) контроля качества, независимый(е) от производства.
19.22 Отдельные работы по проведению контроля, обычно выполняемые отделом(ами) контроля качества могут проводиться в других подразделениях.
19.23 Мероприятия по контролю качества должны включать в себя проведение контроля исходных и упаковочных материалов, а также промежуточных продуктов и АФС.
19.24 Следует проводить оценку любых затруднений, связанных с производством и качеством продукции.
19.25 Маркировка АФС, используемых в клинических исследованиях, должна контролироваться установленным образом и содержать информацию о том, что это вещество предназначено для исследовательских целей.
19.3 Оборудование
19.30 При проведении всех стадий клинических разработок (в т.ч. при использовании опытных участков или лабораторий) для производства АФС, предназначенных для использования в клинических исследованиях, должен быть установлен порядок проведения калибровки и очистки оборудования и методики оценки его соответствия своему назначению.
19.31 Методики по использованию оборудования должны гарантировать, что при работе с материалами риск загрязнения и перекрестного загрязнения минимален.
19.4 Контроль исходных материалов
19.40 Исходные материалы, применяемые для производства АФС, предназначенных для использования в клинических исследованиях, должны оцениваться путем проведения испытаний или поступать с аналитическим паспортом поставщика и проверяться на подлинность. Аналитический паспорт поставщика достаточен для подтверждения качества материала, представляющего опасность.
19.41 В некоторых случаях пригодность исходного материала перед его использованием может быть обоснована не только результатами аналитического контроля, но и путем проведения мелкомасштабных реакций (например, проверкой пригодности).
19.5 Производство
19.50 Ход производства АФС, предназначенных для использования в клинических исследованиях, должен быть зарегистрирован в лабораторных журналах, протоколах серии или другими средствами. Эти документы должны включать в себя информацию об использовании технологических материалов, оборудовании, технологическом процессе и научных наблюдениях.
19.51 Ожидаемые выходы продукции могут различаться более значительно и быть менее определенными, чем ожидаемые выходы продукции процессов, выполняемых в промышленном масштабе. Нет необходимости проводить изучение различия выходов продукции.
19.6 Аттестация (валидация)
19.60 Как правило, не требуется проводить аттестацию (валидацию) процесса производства АФС, предназначенных для использования в клинических исследованиях, если производится одна серия АФС или из-за изменений, вносимых в технологический процесс при разработке АФС, повторение серий является затруднительным или неточным. В этот период разработки качество АФС обеспечивается сочетанием контроля, калибровки (поверки) и аттестации оборудования.
19.61 Аттестацию (валидацию) процесса, в соответствии с разделом 12 настоящего Руководства, следует проводить для серий, предназначенных для реализации, даже если такие серии производятся на опытном или мелкомасштабном производстве.
19.7 Изменения
19.70 Изменения вносятся по мере приобретения новых знаний и роста масштаба производства. Каждое изменение в технологии производства, спецификациях или методиках испытаний должно быть зарегистрировано установленным образом.
19.8 Лабораторный контроль
19.80 Несмотря на то, что аналитические методы, используемые для оценки качества серии АФС, предназначенного для клинических исследований, не могут быть аттестованы (валидированы), они должны быть научно обоснованы.
19.81 Следует иметь систему хранения архивных образцов всех серий, обеспечивающую хранение каждого из архивных образцов в достаточном количестве и надлежащих условиях в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
19.82 Срок годности и дата повторного контроля, определенные в подразделе 11.6 настоящего Руководства, применимы к существующим АФС, предназначенным для использования в клинических исследованиях. Для новых АФС, находящихся на ранних стадиях клинических исследований, положения подраздела 11.6, как правило, применимы.
19.9 Документация
19.90 Система документации должна гарантировать, что информация, полученная в ходе разработки и производства АФС, предназначенных для использования в клинических исследованиях, оформляется документально должным образом и доступна для использования.
19.91 Разработка и применение аналитических методов, которые используются для подтверждения выпуска серии АФС, предназначенной для клинических исследований, должны быть оформлены документально.
19.92 Следует разработать и применять порядок хранения протоколов производства и контроля качества и документации. Этот порядок должен обеспечивать хранение протоколов и документов в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
20 Термины и определения
активная фармацевтическая субстанция (АФС) (active pharmaceutical ingredient (API): Любое вещество или смесь веществ, предназначенные для производства лекарственных средств, которые в производстве лекарственного средства становятся активным ингредиентом этого лекарственного средства. Такие вещества предназначены для проявления фармакологической активности или другого прямого эффекта при диагностике, лечении, облегчении симптомов или профилактике болезни или для воздействия на структуру или функцию организма.
аттестация (qualification): Документальное подтверждение того, что методика, процесс, оборудование, материал или система соответствуют заданным требованиям и их использование дает ожидаемые результаты.
биологическая нагрузка (bioburden): Уровень и тип (например, патогенных или непатогенных) микроорганизмов, которые могут присутствовать в исходных материалах, исходных материалах АФС, промежуточных продуктах или АФС. Биологическая нагрузка не рассматривается как загрязнение, если ее уровни не превышают установленные предельные значения или не происходит обнаружения определенных патогенных микроорганизмов.
валидация (validation): Синоним термина "аттестация".
внутрипроизводственный контроль (или технологический контроль) (in-process control (or-process control): Контроль, выполняемый в ходе технологического процесса с целью проверки соответствия промежуточного продукта или АФС заданным требованиям, по результатам которого могут корректироваться параметры технологического процесса.
вспомогательные средства (process aids): Материалы (за исключением растворителей), используемые как добавки при производстве промежуточного продукта или АФС, которые сами по себе не участвуют в химической или биологической реакции (например, порошок для фильтрования, активированный уголь и т.д.).
выход, ожидаемый (yield, expected): Количество или процент теоретического выхода материала, ожидаемый на любой соответствующей стадии производства, определяемый на основании данных, предварительно полученных при производстве этого материала в лабораторных, опытных или промышленных условиях.
выход, теоретический (yield, theoretical): Количество, которое было бы получено на любой соответствующей стадии производства, рассчитанное на основе количества используемого материала в предполагаемых условиях отсутствия любых потерь или ошибок при фактическом производстве.
дата повторного контроля (retest date): Дата прохождения материалом повторной проверки для подтверждения его пригодности для дальнейшего использования.
загрязнение (contamination): Нежелательное внесение примесей химического или микробиологического происхождения или постороннего материала в исходный материал, промежуточный продукт или АФС в ходе производства, отбора проб, упаковки или переупаковки, хранения или транспортировании.
инструкция, методика, процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования), который прямо или косвенно связан с производством промежуточного продукта или АФС.
исходный материал АФС (API starting material): Сырье, промежуточный продукт или АФС, которые используются при производстве АФС и внедряются в структуру АФС в качестве существенного элемента. Исходный материал АФС может быть продуктом (материалом), получаемым от одного или более поставщиков по контракту (коммерческому соглашению), или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
калибровка (поверка) (calibration): См. общие термины и определения.
карантин (quarantine): Статус материалов, изолированных физически или иным образом на время до вынесения решения об их выпуске или отклонении.
контроль качества (quality control (QC): Проведение проверок или испытаний на соответствие требованиям спецификаций.
критерии приемлемости (acceptance criteria): Численные предельные значения, диапазоны или другие критерии, применимые для приема результатов испытаний.
критический (critical): Определение стадий технологического процесса, технологических условий, требований при проведении испытания, другого существенного параметра или пункта, который следует контролировать в рамках предварительно установленных критериев для гарантии соответствия АФС требованиям своей спецификации.
лекарственная субстанция (drug substance): См. активную фармацевтическую субстанцию.
лекарственный (медицинский) препарат (drug (medicinal) product): Дозированная форма лекарственного средства в первичной окончательной упаковке, предназначенной для продажи.
материал (material): Термин, используемый для определения сырья (исходных материалов, реактивов, растворителей), технологических добавок, промежуточных продуктов, АФС, упаковочных и печатных материалов.
маточный раствор (mother liquor): Остаточная жидкость после процессов кристаллизации и выделения.
Примечание - Маточный раствор может содержать непрореагировавшие материалы, промежуточные продукты, АФС и/или примеси в существенном количестве, а также использоваться для дальнейшей обработки.
методика аттестации (валидации) (validation protocol): Документально оформленный план, устанавливающий порядок аттестации (валидации) и определяющий критерии приемлемости.
Например, методика аттестации (валидации) технологического процесса, конкретно определяющая технологическое оборудование, критические технологические параметры (рабочие диапазоны), характеристики продукции, отбор проб, данные испытаний, которые должны быть собраны, количество циклов аттестации (валидации) и приемлемые результаты испытаний.
номер партии (lot number): См. номер серии.
номер серии (партии) (batch number or lot number): Определенное сочетание цифр, букв и/или символов, обозначающее серию продукции. По номеру серии может быть прослежен ход производства и реализации этой серии.
обеспечение качества (quality assurance (QA): Общая совокупность организационных мероприятий, проводимых с целью обеспечения требуемого качества всех АФС и поддержания систем качества на должном уровне.
отдел(ы) (служба) качества (quality unit(s): Организационное подразделение, независимое от производства, которое несет ответственность за обеспечение и контроль качества, представляющее собой отдельные отделы контроля и обеспечения качества либо одно лицо или группу, в зависимости от размера и структуры организации.
отклонение (deviation): Отступление от утвержденной инструкции или установленного стандарта.
партия (lot): См. серию.
перекрестное загрязнение (cross-contamination): Загрязнение материала или продукта другим материалом или продуктом.
переработка (reworking): Обработка промежуточного продукта или АФС, не удовлетворяющих требованиям стандартов или спецификаций, с помощью одного или нескольких технологических стадий, отличающихся от установленного технологического процесса, с целью получения промежуточного продукта или АФС приемлемого качества (например, перекристаллизация с другим растворителем).
повторная обработка (reprocessing): Повторное введение промежуточного продукта или АФС (в т.ч. не удовлетворяющих требованиям спецификаций) в процесс и повторение стадии кристаллизации или другой соответствующей стадии химической или физической обработки (например, дистилляции, фильтрации, хроматографии. измельчения), которая является частью установленного технологического процесса. Продолжение технологической стадии после того, как внутрипроизводственный контроль показал, что она еще не завершена, считается частью текущего процесса производства и не рассматривается как повторная обработка.
подпись (согласование) (signature (signed): См. согласование.
подрядчик (contract manufacturer): Производитель, выполняющий некоторые производственные операции от имени первоначального производителя.
примесь (impurity): Компонент, присутствующий в промежуточном продукте или АФС, который является нежелательным для этого продукта.
производство (manufacture): Операции и виды контроля, связанные с закупкой исходных материалов, их приемкой, обработкой, упаковкой, выпуском в реализацию, хранением и отгрузкой АФС.
промежуточный продукт (intermediate): Материал, получаемый на стадиях производства АФС, который претерпевает дальнейшие молекулярные изменения или очистку до того, как он может считаться АФС. Промежуточные продукты могут и не выделяться в качестве чистых продуктов.
Примечание - Данное Руководство касается только промежуточных продуктов, производимых со стадии, являющейся (по мнению компании), началом производства АФС.
растворитель (solvent): Неорганическая или органическая жидкость, используемая в качестве носителя для приготовления растворов или суспензий при производстве промежуточного продукта или АФС.
серия (партия) (batch or lot): Определенное количество материалов, произведенных в одном или нескольких технологических процессах так, что их можно считать однородными в рамках установленных пределов. При непрерывном производстве понятие серии может относиться к определенной части продукции, характеризуемой однородностью. Размер серии может быть установлен по фиксированному количеству продукции либо по количеству продукции, производимой за установленный период времени (для целей настоящего приложения).
система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
согласование (подпись) (signed signature): Подпись лица, выполняющего конкретное действие или обзор, в виде сокращенной или полной фамилии и инициалов, сделанной от руки, личной печати или авторизованной и защищенной электронной подписи.
состав примесей (impurity profile): Описание идентифицированных и неидентифицированных примесей, присутствующих в АФС.
спецификация (specification): Перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, устанавливающие численные границы, диапазоны или другие критерии для указанных испытаний. Спецификация устанавливает критерии, которым должен соответствовать материал для того, чтобы его можно было применять по назначению. Считается, что материал соответствует требованиям спецификации, если при испытаниях по принятым аналитическим методикам он удовлетворяет заданным критериям приемлемости (для целей настоящего приложения. См. также общие термины и определения).
срок годности (expiry date or expiration date): Дата, помещаемая на упаковку/этикетку АФС, обозначающая период времени, в течение которого гарантируется сохранение свойств АФС в рамках установленных спецификаций при хранении в определенных условиях и после которого данная АФС не должна использоваться.
сырье (raw material): Общий термин, используемый для обозначения исходных материалов, реактивов и растворителей, предназначенных для производства промежуточного продукта или АФС.
технологический контроль (process control): См. внутрипроизводственный контроль.
технологический процесс (production): Операции, включающие в себя приемку исходных материалов, их обработку, упаковку и получение готовой АФС (для целей настоящего приложения).
упаковочный материал (packaging material): Любой материал, предназначенный для защиты промежуточного продукта или АФС при хранении и транспортировании (для целей настоящего приложения).
эталонный стандарт, вторичный (reference standard, secondary): Вещество установленного качества и чистоты по результатам сравнения с первичным эталонным стандартом, используемое в качестве стандарта сравнения для текущих лабораторных анализов.
эталонный стандарт, первичный (reference standard, primary): Вещество, признанное подлинным материалом высокой степени чистоты по результатам обширных аналитических испытаний. Этот стандарт может быть: 1) получен из официально признанного источника; 2) приготовлен путем независимого синтеза; 3) получен из имеющегося промышленного материала высокой степени чистоты или 4) приготовлен путем дальнейшей очистки существующего промышленного материала.
Общие термины и определения
В настоящем стандарте используются следующие термины с соответствующими определениями:
1 аттестация (валидация) (qualification, validation): Доказательство того, что методика, процесс, оборудование, материал, операция или система соответствуют заданным требованиям и их использование действительно дает ожидаемые результаты.
2 баллон (cylinder): Сосуд для хранения газа при высоком давлении.
3 банки клеток (cell bank)
система банков клеток (cell bank system): Система, при которой последовательные серии продукции производят из клеточных культур, принадлежащих главному банку клеток, который полностью охарактеризован на подлинность и отсутствие загрязнений. Некоторое количество емкостей главного банка клеток используется для формирования рабочего банка клеток. Система банков клеток аттестуется (валидируется) на определенное количество пересевов или количество удвоений популяции, до достижения которых они могут использоваться в текущем производстве.
главный банк клеток (master cell bank): Культура клеток, полностью охарактеризованная на подлинность и отсутствие загрязнений, распределенная по емкостям в процессе одной операции таким образом, чтобы обеспечивались ее однородность и стабильность. Как правило, главный банк клеток хранится при температуре минус 70 °С или ниже.
рабочий банк клеток (working cell bank): Культура клеток, взятая из главного банка клеток для приготовления производственных культур клеток. Рабочие банки клеток хранят при температуре минус 70 °С или ниже.
4 биологические агенты (biological agents): Микроорганизмы, в т.ч. полученные методами генной инженерии, культуры клеток и эндопаразиты, патогенные или непатогенные.
5 биореактор (biogenerator): Изолированная система (например, ферментатор), в которую вместе с другими материалами вводят биологические агенты и в результате протекающей реакции происходит их размножение или образование других веществ. Биореакторы, как правило, оборудуются устройствами для регулирования, контроля, добавления и извлечения материалов.
6 валидация (validation): См. аттестация.
7 внутрипроизводственный контроль (in-process control): Контроль, выполняемый в ходе технологического процесса с целью проверки соответствия продукции заданным требованиям, по результатам которого может выполняться корректировка параметров технологического процесса. Контроль состояния окружающей среды или оборудования рассматривается как элемент внутрипроизводственного контроля.
8 возврат (return): Возврат лекарственного средства его производителю или поставщику.
9 воздушный шлюз (air-lock): Ограниченное пространство с двумя или несколькими дверями между двумя или несколькими помещениями (например, различных классов чистоты), предназначенное для разделения воздушных сред помещений при входе в них. Воздушный шлюз служит для перехода персонала или перемещения материалов.
10 готовая продукция (готовый продукт) (finished product): Лекарственное средство, прошедшее все этапы технологического процесса, в т.ч. окончательную упаковку.
11 изолированная зона (contained area): Зона, оборудованная соответствующими фильтрами и устройствами подготовки воздуха для предотвращения загрязнения внешней окружающей среды биологическими агентами, присутствующими в этой зоне.
12 изоляция (containment): Меры по ограничению распространения биологического агента или другого вещества за пределы определенного пространства.
первичная изоляция: Изоляция, препятствующая прониканию биологического агента в окружающую среду, непосредственно прилегающую к рабочей зоне. Предусматривает использование закрытых контейнеров или безопасных биологических шкафов и методов безопасного выполнения технологических операций.
вторичная изоляция: Изоляция, препятствующая прониканию биологических агентов во внешнюю окружающую среду или в другие рабочие зоны. Предусматривает использование помещений со специальными системами подготовки воздуха, воздушных шлюзов и/или стерилизаторов для перемещения материалов и обеспечивает безопасное выполнение технологических операций. Во многих случаях используется для повышения эффективности первичной изоляции.
13 инструкция, методика, процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования).
14 инфицированный (infected): Зараженный чужеродными биологическими агентами и способный к распространению инфекции.
15 исходные материалы (starting material): Любое вещество, используемое для производства лекарственных средств, кроме упаковочных материалов.
16 калибровка (поверка) (calibration): Операции, устанавливающие при определенных условиях зависимость между значениями, регистрируемыми контрольно-измерительными приборами (системами) и соответствующими стандартными величинами (эталонами).
17 карантин (quarantine): Статус исходных или упаковочных материалов, промежуточной, нерасфасованной или готовой продукции, изолированных физически или иным образом до вынесения решения об их выпуске или отклонении.
18 коллектор (manifold): Устройство или оборудование, позволяющее одновременно наполнять газом несколько баллонов (контейнеров) от одного источника.
19 контролируемая зона (controlled area): Зона, построенная и эксплуатируемая таким образом, чтобы предотвратить внесение возможного загрязнения и случайное распространение живых организмов (например, может иметь систему воздухоподготовки, соответствующую зоне D). Степень контроля зависит от свойств организмов, используемых в технологическом процессе. Как минимум, такая зона должна иметь отрицательное давление по отношению к смежным помещениям и возможность эффективно удалять незначительные количества аэрозольных загрязнений.
20 контроль качества (quality control): См. пункт 1.4 раздела 1.
21 криогенный сосуд (cryogenic vessel): Сосуд для хранения сжиженных газов при сверхнизких температурах.
22 культура клеток (cell culture): Клеточная масса, полученная в результате выращивания in vitro клеток, изолированных от многоклеточных организмов.
23 лекарственное растение (medicinal plant): Растение или часть его, используемое для медицинских целей.
24 лекарственное средство (medicinal product): Любое вещество или комбинация веществ, предназначенное для лечения или профилактики заболеваний у человека или животных. Любое вещество или комбинация веществ, вводимое человеку или животным с диагностическими целями для восстановления, корректировки или изменения физиологических функций человека или животных, также рассматривается как лекарственное средство.
25 лекарственное средство из растительного сырья (herbal medicinal product): Лекарственное средство, содержащее в качестве активных ингредиентов только вещества растительного происхождения и/или препараты на их основе.
26 нерасфасованный готовый продукт (балк-продукт) (bulk product): Продукт, прошедший все производственные стадии, за исключением окончательной упаковки.
27 номер серии (партии) (batch number or lot number): Определенное сочетание цифр и/или букв, обозначающее серию продукции.
28 перекрестное загрязнение (cross contamination): Загрязнение материалов и продукции другими материалами или продукцией.
29 переработка (reprocessing): Повторная обработка серии или части серии продукции, не соответствующей заданным требованиям, начиная с определенной стадии производства, для получения продукции требуемого качества после проведения одной или нескольких дополнительных операций.
30 повторное использование (recovery): Включение произведенной ранее серии продукции требуемого качества (или ее части) в другую серию продукции на определенной стадии производства.
31 посевной материал (seed lot)
система посевных материалов (seed lot system): Система, при которой последовательные серии продукции получают из главного посевного материала при определенном количестве пересевов (пассажей). В текущем производстве рабочий посевной материал готовится из главного посевного материала. Готовый продукт производится из рабочего посевного материала, при этом число пересевов из главного посевного материала не должно превышать значения, установленного при клинических испытаниях вакцин, исходя из требований безопасности и эффективности. Происхождение главного посевного материала и история пересевов из него должны оформляться документально.
главный посевной материал (master seed lot): Культура микроорганизмов, распределенная из одного нерасфасованного продукта по емкостям в процессе одной операции таким образом, чтобы обеспечивались однородность, стабильность и не допускалось загрязнение. Главный посевной материал в жидком виде хранят при температуре минус 70 °С; в лиофилизированном виде - при температуре, обеспечивающей его стабильность.
рабочий посевной материал (working seed lot): Культура микроорганизмов, полученная из главного посевного материала и предназначенная для использования в производстве. Рабочий посевной материал распределяется по емкостям и хранится аналогично хранению главного посевного материала.
32 производитель (manufacturer): Держатель лицензии на производство.
Примечание - Предприятие, осуществляющее хотя бы один этап производства, рассматривается как производитель лекарственных средств.
33 производство (manufacture): Все операции и виды контроля, связанные с получением, приемкой и обработкой исходных материалов, упаковкой, выпуском в реализацию, хранением и отгрузкой лекарственных средств.
34 промежуточный продукт (intermediate product): Частично обработанный материал, который должен пройти дальнейшие стадии производства, прежде чем он станет нерасфасованным готовым продуктом.
35 промышленный регламент, технологические инструкции и инструкции по упаковке (manufacturing formulae, processing and packaging instructions): Документы, определяющие все используемые исходные материалы и операции по производству и упаковке продукции.
Примечания
1 Промышленный регламент является технологическим документом действующего серийного производства, устанавливающим методы производства, технологические нормативы, средства, условия и порядок проведения технологического процесса и обеспечивающим получение лекарственного средства с показателями качества, отвечающими требованиям нормативной документации.
2 Данный термин используется только в области производства лекарственных средств.
3 Промышленный регламент не относится к техническим регламентам, предусмотренным Федеральным законом "О техническом регулировании".
36 протокол на серию (record): Документ, отражающий ход производства каждой серии продукции, в т.ч. разрешение на ее реализацию, и все факторы, имеющие отношение к качеству готовой продукции.
37 радиофармацевтический препарат (radiopharmaceutical): Любое лекарственное средство, содержащее в готовом виде один или более радионуклидов (радиоактивных изотопов), используемых для медицинских целей.
38 растительное сырье (crude plant, vegetable drug): Сырые или высушенные лекарственные растения или их части.
39 регистрационное досье (registration dossier): Комплект документов и материалов установленной структуры и содержания, представляемый вместе с заявкой на регистрацию лекарственного средства, а также утвержденный в процессе регистрации.
40 серия (партия) (batch, or lot): Определенное количество однородных исходных и упаковочных материалов или однородной продукции, обработанной в ходе одной или нескольких последовательных технологических стадий.
Примечание - При необходимости на определенных стадиях производства серия может быть разделена на подсерии, объединяемые впоследствии в однородную серию продукции. При непрерывном производстве понятие серии должно относиться к определенной части продукции, характеризуемой однородностью.
С точки зрения контроля готовой продукции серия продукции включает в себя совокупность единиц дозированной формы лекарственных средств (лекарственной формы), изготовленных из одного объема исходного материала и прошедших единую последовательность производственных операций или единый цикл стерилизации; при непрерывном производстве - все единицы, произведенные в заданный интервал времени.
41 сжиженные газы (liquifiable gases): Газы, которые при стандартных температуре и давлении наполнения находятся в баллоне в сжиженном виде.
42 система (system): Совокупность взаимосвязанных действий и технических средств, образующих единое целое.
43 система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
44 сопоставление (выход продукции) (reconciliation): Сравнение ожидаемого и фактического объемов произведенной или использованной продукции с учетом стандартных отклонений.
45 спецификация (specification): Документ, содержащий требования, предъявляемые к материалам и продуктам, используемым или получаемым при производстве, являющийся основой для оценки качества лекарственных средств.
46 стерильность (sterility): Отсутствие живых микроорганизмов. Требования к проведению контроля стерильности приведены в соответствующей нормативной документации.
47 технологический процесс (production): Операции, включающие в себя приемку и обработку исходных материалов, упаковку и получение готового продукта.
48 упаковка (packaging): Все операции, в т.ч. наполнение и маркировка, проводимые с нерасфасованным продуктом для получения готового продукта.
Примечание - Стерильное наполнение, как правило, не следует рассматривать как часть процесса упаковки. В этом случае нерасфасованным продуктом считаются наполненные первичные контейнеры без окончательной упаковки.
49 упаковочный материал (packaging material): Любой материал, применяемый для упаковывания лекарственных средств, за исключением внешней упаковки, используемой для транспортирования. Упаковочные материалы делятся на первичные или вторичные в зависимости от наличия прямого контакта с продуктом.
50 Уполномоченное лицо (Qualified person): Сотрудник предприятия-производителя, принимающий окончательное решение о выпуске серии лекарственного средства.
51 чистая зона (clean area): Зона, построенная и эксплуатируемая таким образом, что в ней сведено к минимуму проникание, образование и накопление загрязнений в виде частиц и микроорганизмов.
Примечание - Типы чистых зон определены в приложении 1.
52 чистая изолированная зона (clean contained area): Зона, построенная и эксплуатируемая таким образом, что она одновременно является чистой и изолированной зоной.
53 экзотический организм (exotic organism): Биологический агент, вызывающий заболевание, отсутствующее в данной стране или географической зоне, либо являющееся объектом профилактических мер или программы по его устранению.
Текст документа сверен по:
официальное издание
М.: ИПК Издательство стандартов, 2004