ГОСТ Р ИСО 14160-2003
Группа Р26
ГОСУДАРСТВЕННЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
СТЕРИЛИЗАЦИЯ ОДНОРАЗОВЫХ МЕДИЦИНСКИХ ИЗДЕЛИЙ,
СОДЕРЖАЩИХ МАТЕРИАЛЫ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ
Валидация и текущий контроль стерилизации
с помощью жидких стерилизующих средств
Sterilization of single-use medical devices incorporating materials of animal origin.
Validation and routine control of sterilization by liquid sterilants
ОКС 11.080
ОКП 94 5120
Дата введения 2004-01-01
Предисловие
1 ПОДГОТОВЛЕН Ассоциацией инженеров по контролю микрозагрязнений (АСИНКОМ)
ВНЕСЕН Техническим комитетом по стандартизации ТК 383 "Стерилизация медицинских изделий"
2 ПРИНЯТ И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Госстандарта России от 16 января 2003 г. N 8-ст
3 Настоящий стандарт представляет собой аутентичный текст международного стандарта ИСО 14160-98 "Стерилизация одноразовых медицинских изделий, содержащих материалы животного происхождения. Валидация и текущий контроль стерилизации с помощью диких стерилизующих средств"
4 ВВЕДЕН ВПЕРВЫЕ
Введение
Стерильным является изделие, свободное от жизнеспособных микроорганизмов. При поставке стерильного изделия международные стандарты требуют, чтобы до начала стерилизации случайная контаминация (загрязнение) медицинских изделий от всех источников была сведена к минимуму всеми практически возможными способами. Тем не менее, даже при производстве медицинских изделий с соблюдением требований системы качества (ИСО 13485 и ИСО 13488) до начала стерилизации на них могут быть микроорганизмы, хотя и в небольшом количестве. Такие изделия являются нестерильными. Целью процесса стерилизации является инактивация микробиологических контаминантов и таким образом превращение нестерильных предметов в стерильные.
Инактивация чистой культуры микроорганизмов физическими и/или химическими агентами, используемыми для стерилизации медицинских изделий, приближенно описывается экспоненциальным законом. Это означает, что независимо от примененной степени обработки существует конечная вероятность того, что микроорганизм может выжить. При конкретной обработке вероятность выживания определяется числом и типами микроорганизмов и окружающими условиями, в которых микроорганизмы находятся при обработке, т.е. стерильность любого отдельного изделия из группы подвергаемых стерилизации изделий не может быть гарантирована, и стерильность обработанной группы изделий должна выражаться через вероятность присутствия на изделии жизнеспособного микроорганизма.
Требования к системе качества при проектировании, разработке, производстве, монтаже и обслуживании приведены в стандартах серии ИСО 9000, а также ИСО 13485 и ИСО 13488. Стандарты серии ИСО 9000 определяют некоторые процессы, используемые при производстве, как специальные, если их результаты не могут быть полностью подтверждены последующим контролем и испытанием продукции. Стерилизация является примером специального процесса, поскольку эффективность ее не может быть подтверждена контролем и испытанием продукции. Поэтому до ввода оборудования в эксплуатацию следует выполнить валидацию процессов стерилизации, а во время эксплуатации проводить текущий контроль и техническое обслуживание оборудования.
Важно сознавать, что должным образом валидированный и точно контролируемый процесс стерилизации не является единственным фактором, обусловливающим уверенность в том, что продукция является стерильной и пригодной для предназначенного использования. Необходимо также обратить внимание на целый ряд факторов, включающих микробиологический статус (бионагрузку) поступающего сырья и/или компонентов, их последующее хранение, а также контроль среды, в которой производится, собирается и упаковывается продукция.
Для стерилизации медицинских изделий наиболее часто используют влажное тепло, сухое тепло (нагретый воздух), ионизирующее излучение и оксид этилена. В то время как некоторые изделия, содержащие ткани животных, могут быть совместимы с общепринятыми методами стерилизации (например, нити из кетгута обычно стерилизуются облучением), другие изделия (биологические сердечные клапаны или тканевые прокладки) не совместимы с этими процессами стерилизации и могут потребоваться другие стерилизующие агенты. В таких случаях широко используются жидкие химические стерилизующие средства, причем как и при использовании других методов стерилизации, эффективность процесса необходимо подтвердить и зафиксировать перед тем, как принять его для повседневного использования.
1 Область применения
Настоящий стандарт устанавливает требования к разработке, валидации, управлению и контролю процесса стерилизации жидкими стерилизующими средствами одноразовых медицинских изделий, состоящих полностью или частично из материалов животного происхождения.
Настоящий стандарт не распространяется на материалы человеческого происхождения, не описывает систему обеспечения качества при контроле всех стадий производства и не рассматривает методы валидации способов инактивации вирусов.
Примечания
1. Стандарты на системы качества (ГОСТ Р ИСО 9001 и ГОСТ Р 51536, или ГОСТ Р ИСО 9002 и ГОСТ Р 51537) могут быть использованы для контроля всех стадий производства, включая процесс стерилизации.
2 При разработке технологии обработки медицинских изделий, содержащих материалы животного происхождения, необходимо рассматривать эффективность воздействия жидких химических стерилизующих средств на возможные вирусные контаминанты с учетом происхождения материалов, используемых при производстве этих конкретных медицинских изделий. Хотя важность вопроса валидации вирусной инактивации в процессах, составляющих область применения настоящего стандарта, очевидна, этот аспект из него исключен; в разработке находится отдельный Европейский Стандарт [1].
3 Жидкие химические стерилизующие средства, традиционно используемые для стерилизации животных тканей в медицинских изделиях, могут быть неэффективны для инактивации патогенных агентов трансмиссивных губчатых форм энцефалопатий, таких как губчатая форма бычьей энцефалопатии (BSE) или скреппи (возбудитель инфекционного вирусного заболевания взрослых овец). Удовлетворительную валидацию в соответствии с настоящим стандартом нельзя рассматривать как подтверждение инактивации инфицирующих агентов этого типа.
Руководство по оценке соответствия настоящему стандарту приведено в приложении А.
2 Нормативные ссылки
В настоящем стандарте использованы ссылки на следующие стандарты:
ГОСТ Р ИСО 9001-96 Системы качества. Модель обеспечения качества при проектировании, разработке, производстве, монтаже и обслуживании.
ГОСТ Р ИСО 9001-2001 Системы менеджмента качества. Требования.
ГОСТ Р ИСО 9002-96 Системы качества. Модель обеспечения качества при производстве, монтаже и обслуживания
ГОСТ Р ИСО 9004-2001 Система менеджмента качества. Руководство по улучшению деятельности
ГОСТ Р ИСО 11134-2000 Требования к валидации и текущему контролю. Промышленная стерилизация влажным теплом
ГОСТ Р ИСО 11135-2000 Медицинские изделия. Валидация и текущий контроль стерилизации оксидом этилена
ГОСТ Р ИСО 11137-2000 Стерилизация медицинской продукции. Требования к валидации и текущему контролю. Радиационная стерилизация
ГОСТ Р ИСО 11138-1-2000 Стерилизация медицинской продукции. Биологические индикаторы. Часть 1. Общие требования
ГОСТ Р ИСО 11737-1-2000 Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукции
ГОСТ Р ИСО 13408-1-2000 Асептическое производство медицинской продукции. Общие требования
ГОСТ Р 51536-99 (ИСО 13485-96) Системы качества. Медицинские изделия. Частные требования к применению ГОСТ Р ИСО 9001
ГОСТ Р 51537-99 (ИСО 13488-96) Системы качества. Медицинские изделия. Частные требования к применению ГОСТ Р ИСО 9002
Примечание - Взаимосвязь международных и европейских стандартов приведена в приложении В.
3 Определения
В настоящем стандарте приняты следующие термины с соответствующими определениями:
3.1 партия, серия (batch): Количество нерасфасованной, промежуточной или готовой продукции, свойства и качество которой предполагаются однородными и которая изготовлена в течение определенного цикла производства.
3.2 бионагрузка (bioburden): Популяция жизнеспособных микроорганизмов на/в продукции и/или упаковке.
3.3 носитель (carrier): Удерживающий материал, на который нанесены тест-микроорганизмы.
3.4 комиссионная приемка (commissioning): Получение и документирование данных о том, что оборудование поставлено и смонтировано в соответствии с технической документацией и при соблюдении инструкций по эксплуатации оно функционирует в предусмотренных пределах.
3.5 величина десятикратного сокращения (decimal reduction value); величина D (D-value): Время выдержки или поглощенная доза излучения, необходимые для уменьшения популяции тест-микроорганизмов в 10 раз при определенных условиях обработки.
3.6 время выдержки (exposure time): Время, в течение которого медицинское изделие подвергается воздействию определенной температуры и стерилизующего вещества в определенной концентрации.
3.7 инактивация (inactivation): Потеря тест-микроорганизмами способности к росту, прорастанию и/или размножению при определенных условиях культивирования.
Примечание - В настоящем стандарте под тест-микроорганизмами подразумеваются спорообразующие и неспорообразующие бактерии, вирусы, грибы и простейшие.
3.8 инокулированный носитель (inoculated carrier): Носитель, на который нанесено определенное количество тест-микроорганизмов.
3.9 жидкое химическое стерилизующее средство (liquid chemical sterilant): Определенная композиция химических веществ в виде раствора или в жидкой форме, которая применяется для стерилизации.
3.10 медицинское изделие (medical device): Инструмент, аппарат, приспособление, материал, используемые отдельно или с другими изделиями, необходимым программным обеспечением, предназначенные для людей в целях:
- диагностики, профилактики, наблюдения, лечения или облегчения болезни;
- диагностики, наблюдения, лечения, облегчения или компенсации при травмах или инвалидности;
- исследования, замещения или изменения анатомии или физиологического процесса;
- контроля зачатия,
основное действие которых внутри или снаружи тела человека достигается без применения фармакологических, иммунологических или метаболических средств, но которые могут применяться совместно с ними.
3.11 аттестация эксплуатируемого оборудования (performance qualification): Получение и документирование данных о том, что при выполнении требований технологии процесса оборудование после комиссионной приемки будет производить надлежащую продукцию.
3.12 предстерилизационное число (presterilization count): Количество жизнеспособных микроорганизмов, обнаруженное перед стерилизацией.
3.13 совместимость продукции (product compatibility): Способность процесса стерилизации обеспечить намеченные результаты без ущерба для продукции.
3.14 разработка процесса (process development): Документированная программа исследований, проводимых с целью определения параметров процесса стерилизации с учетом свойств продукции, упаковки, схемы загрузки и/или ограничений, накладываемых характеристиками оборудования.
3.15 ревалидация (revalidation): Частное или полное повторение валидации для подтверждения надежности процесса.
3.16 стерильность (sterility): Отсутствие жизнеспособных микроорганизмов.
Примечание - На практике состояние абсолютного отсутствия.
3.17 стерильный (sterile): He содержащий жизнеспособных микроорганизмов.
Примечание - На практике состояние абсолютного отсутствия микроорганизмов не может быть доказано.
3.18 стерилизация (sterilization): Валидированный процесс освобождения продукта от жизнеспособных микроорганизмов всех форм.
Примечание - При стерилизации процесс отмирания микроорганизмов описывается экспоненциальным законом. Следовательно, наличие микроорганизмов в конкретном объекте может быть выражено в вероятностном виде. Хотя эта вероятность может быть снижена до весьма малой величины, она никогда не может быть снижена до нуля. Вероятность может быть выражена через уровень обеспечения стерильности УС (sterility assurance level - SAL), обычно выражаемый в форме 10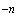 .
.
3.19 консервирующий раствор (storage solution): Жидкость, в которой медицинское изделие в его конечной форме подается для использования.
3.20 валидация (validation): Документированная процедура получения, протоколирования и интерпретации результатов, необходимых для демонстрации того, что процесс неизменно дает продукт, соответствующий предварительно определенным требованиям.
Примечание - Применительно к стерилизации жидкими химическими стерилизующими средствами под валидацией понимается общая программа, состоящая из комиссионной приемки и аттестации эксплуатируемого оборудования.
3.21 число живых микроорганизмов (viable count): Число микроорганизмов, определяемое по росту отдельных колоний при установленных условиях культивирования.
Примечание - Отдельная колония не обязательно образуется от единичного жизнеспособного микроорганизма.
4 Общие положения
4.1 Контроль производства
Чтобы предстерилизационное число находилось в установленных пределах, следует разработать и контролировать производственный процесс.
Примечание - Этим требованиям отвечает использование системы качества в соответствии с ГОСТ Р 51536 или ГОСТ Р 51537.
Для контроля источников сырья животного происхождения должна быть разработана и использована документированная система.
Примечание - Разрабатывается Европейский Стандарт по источникам, контролю, сбору материала и обращению с ним [2].
Документация и протоколы должны пересматриваться и утверждаться ответственным персоналом (4.2).
4.2 Персонал
Ответственность за обслуживание оборудования (4.4), валидацию (раздел 5) и текущий контроль (раздел 6) стерилизации путем выдерживания в жидких химических стерилизующих средствах, а также за выпуск продукции должна быть возложена на квалифицированный персонал, как указано в ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002.
4.3 Калибровка
Должна быть разработана, документирована и утверждена эффективная система калибровки всех контролирующих, показывающих и записывающих приборов, используемых при валидации и текущем контроле процесса стерилизации. Эта система должна соответствовать требованиям ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002.
4.4 Техническое обслуживание оборудования
Техническое обслуживание оборудования должно планироваться и осуществляться в соответствии с документированными процедурами. Процедура для каждого планового задания по обслуживанию и периодичность должны быть определены и документированы.
Оборудование для обработки медицинских изделий не должно использоваться до тех пор, пока все задания по техническому обслуживанию не будут успешно завершены и документированы.
Протоколы обслуживания должны храниться в соответствии с требованиями ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002.
Процедуры и протоколы обслуживания должны периодически пересматриваться ответственным лицом (4.2).
4.5 Разработка процесса и совместимость продукции
4.5.1 Перед началом работы с новой или модернизированной продукцией, упаковкой, схемой загрузки или процессом стерилизации следует определить и документально оформить процесс стерилизации, подлежащий валидации.
При этом следует показать эквивалентность ранее валидированному процессу новых решений для продукции, упаковки или схемы загрузки.
Доказательство эквивалентности следует оформить документально.
Примечание - Указанный процесс стерилизации может включать раздельную обработку более чем одним жидким химическим стерилизующим средством.
4.5.2 Конструкция продукта и упаковки должны обеспечивать их контакт с жидким химическим стерилизующим средством, при этом остатки последнего после стерилизации должны быть ниже уровня, указанного производителем. Должны быть указаны места внутри продукции, наиболее трудные для проведения стерилизации.
4.5.3 Должно быть показано и документировано, что процесс стерилизации не ухудшает потребительские свойства продукции или ее упаковки. При необходимости повторной стерилизации ее влияние должно быть оценено и документировано.
5 Валидация
5.1 Общие положения
Процедуры валидации должны быть документированы, а протоколы каждой валидации должны храниться (5.4.1).
5.2 Комиссионная приемка
Комиссионная приемка должна подтвердить, что технические характеристики оборудования, используемого для процесса стерилизации, соответствуют требованиям.
5.3 Аттестация эксплуатируемого оборудования
5.3.1 Аттестация эксплуатируемого оборудования должна показать, что процесс стерилизации:
a) обладает соответствующей летальной активностью против представительной группы микроорганизмов (5.3.5, 5.3.6 и А.5);
b) характеризуется определенными технологическими параметрами (температурой, концентрацией жидкого химического стерилизующего средства, рН и т.п.), которые могут контролироваться в процессе стерилизации.
5.3.2 При проведении аттестации эксплуатируемого оборудования часть продукции, наиболее трудная для стерилизации (4.5.2), должна быть предметом особого внимания.
5.3.3 Предстерилизационное число должно соответствовать требованиям ГОСТ Р ИСО 11737-1.
5.3.4 Прежде чем начать аттестацию эксплуатируемого оборудования, должна быть валидирована стадия нейтрализации жидкого химического стерилизующего средства, которая предшествует культивированию выживших микроорганизмов. Метод нейтрализации не должен вносить ошибку в расшифровку и объяснение результатов культивирования.
5.3.5 В рамках технологии процесса должно быть указано сочетание условий с наименьшей бактерицидной активностью, которое должно использоваться при аттестации эксплуатируемого оборудования.
5.3.6 Микробиологическая аттестация эксплуатируемого оборудования должна включать три этапа:
a) проведение исследований с целью определения микроорганизмов, обладающих высокой резистентностью к данному процессу стерилизации (А.4.2.3);
b) определение кинетики их инактивации.
На этом этапе строят логарифмическую кривую выживания для микроорганизмов, наиболее резистентных к данному процессу. Кривую выживания строят не менее чем по пяти точкам, охватывающим, по меньше мере, тысячекратное снижение числа микроорганизмов (А.4.2.4.1 и А.4.2.5). Если продукция не позволяет провести такую процедуру построения логарифмической кривой выживания, должен использоваться метод MPN (наиболее вероятного числа НВЧ - most probable number), указанный в А.4.2.4.2. Эту замену следует обосновать и документировать.
Микроорганизмы для испытаний необходимо наносить на материал носителя, который представляет материал медицинского изделия;
c) оценку инактивации микроорганизмов относительно предстерилизационного числа по их прорастанию на носителе из тканей.
В дополнение к изолятам, выделенным из бионагрузки, виды используемых микроорганизмов должны включать микроорганизмы с известной высокой резистентностью к данному процессу стерилизации и, в любом случае, с резистентностью, эквивалентной резистентности спор Bacillus subtilis (таблица А.2) в соответствии с ГОСТ Р ИСО 11138-1.
Примечание - Пли планировании таких экспериментов необходимо учитывать уровень органической и неорганической контаминации и вариацию между повторностями в экспериментах.
5.3.7 В процессе стерилизации время выдержки должно быть не менее  , где
, где 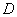 - величина наиболее резистентного микроорганизма, определенная при аттестации эксплуатируемого оборудования, а
- величина наиболее резистентного микроорганизма, определенная при аттестации эксплуатируемого оборудования, а 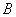 - бионагрузка, определенная по ГОСТ Р ИСО 11737-1.
- бионагрузка, определенная по ГОСТ Р ИСО 11737-1.
Примечание - Увеличенная длительность обработки обеспечивает эффективность стерилизации, которая характеризуется вероятностью выживших микроорганизмов (уровнем стерильности) не более 10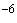 .
.
В соответствии с [1] этот уровень обеспечения стерильности является обязательным для изделий, подвергаемых финишной стерилизации и маркируемых как стерильные (приложение С).
5.3.8 Если после завершения процесса стерилизации медицинское изделие подвергается асептическому перемещению, то необходимо учитывать следующее:
a) процессы, используемые при стерилизации компонентов производства (например, контейнеров, консервирующих растворов), должны валидироваться и подвергаться текущему контролю согласно соответствующему международному стандарту;
b) процедуры перемещения изделий после выдержки в жидком химическом стерилизующем средстве должны быть валидированы в соответствии с ГОСТ Р ИСО 13408-1 (А.4.2.7.).
5.4 Валидация
5.4.1 Протокол валидации, содержащий результаты всех действий, должен быть подписан лицами, ответственными за подготовку, рассмотрение и принятие протокола. Протокол валидации должен храниться по ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002.
5.4.2 Протокол валидации должен иметь ссылку на технологию процесса стерилизации жидким химическим стерилизующим средством. В протоколе должно быть указано медицинское изделие, для которого проводилась валидация, а также следующие данные:
a) частота и метод(ы) определения бионагрузки и область применения результатов;
b) характеристики окружающей среды, в которой было изготовлено жидкое химическое стерилизующее средство и контейнеры, а также осуществлено асептическое перемещение (5.3.8);
c) критерии обучения и сертификации для допуска персонала, ответственного за асептическое перемещение (5.3.8);
d) метод подтверждения отсутствия жизнеспособных микроорганизмов в жидких химических стерилизующих средствах (А. 6);
e) состав жидких химических стерилизующих средств, включая характеристику их компонентов;
f) рН жидкого химического стерилизующего средства;
g) остаточная активность жидкого химического стерилизующего средства после завершения процесса стерилизации в виде концентрации и/или бактерицидной активности;
h) характеристики сосуда для обработки, в котором продукция контактирует с жидким химическим стерилизующим средством, включая конструкционные материалы, размеры и подробности применяемой предварительной обработки;
i) количество продукции, подлежащее стерилизации, на единицу объема жидкого химического стерилизующего средства;
j) время выдержки;
k) температура стерилизации;
l) любые другие критические параметры процесса, выявленные в ходе разработки процесса;
m) метод стерилизации любого консервирующего раствора, в котором продукт поставляется после стерилизации (5.3.8).
5.4.3 В тех случаях, когда валидация конкретного изделия оценивается как пригодная для других изделий, доказательства этой оценки должны быть документированы.
5.5 Ревалидация
5.5.1 Данные валидации и любой последующей ревалидации должны пересматриваться не менее одного раза в год. Должно быть также подготовлено обоснование необходимости ревалидации.
Ревалидация должна предприниматься, если не будут получены достаточные данные, свидетельствующие о приемлемости процесса стерилизации. Процедуры по пересмотру данных валидации и ревалидации должны быть документированы, а протоколы ревалидации должны храниться.
5.5.2 Протокол ревалидации должен быть подписан лицами, назначенными теми же организациями, которые готовили, рассматривали и принимали первоначальный протокол валидации (5.4.1).
6 Контроль процесса
6.1 Бионагрузку следует оценивать по ГОСТ Р ИСО 11737-1 через определенные интервалы времени. Если при проведении текущей оценки предстерилизационного числа выделен микроорганизм, который не был изучен при первоначальной аттестации эксплуатируемого оборудования, то для него должны быть выполнены требования 5.3.5.
6.2 Чтобы убедиться в выполнении технологических требований процесса стерилизации, для каждой партии стерилизуемой продукции необходимо записывать и хранить следующие сведения:
a) переменные величины, контролируемые в ходе стерилизации конечных контейнеров, если это имеет место;
b) переменные величины, контролируемые в ходе стерилизации консервирующего раствора, если это имеет место;
c) исходную концентрацию и рН жидкого химического стерилизующего средства;
d) параметры, контролируемые в ходе приготовления жидкого химического стерилизующего средства;
e) результаты проверки целостности всех фильтров, используемых для стерилизации растворов, если они применяются;
f) время выдержки;
g) температуру выдержки;
h) результаты контроля окружающей среды во время асептического перемещения, если оно применяется;
i) фамилии лиц, которые:
1) готовят консервирующие растворы и растворы жидкого химического стерилизующего средства;
2) контролируют процесс стерилизации;
3) осуществляют асептическое перемещение, если оно применяется;
j) количество обработанной продукции в экземплярах (или других единицах).
6.3 Для каждой партии медицинских изделий, подвергнутых жидкостной химической стерилизации, необходимо исследовать на присутствие жизнеспособных микроорганизмов следующие объекты:
a) химические стерилизующие растворы;
b) консервирующие растворы, если они применяются;
c) по крайней мере, одно из перечисленного ниже:
1) конечный продукт;
2) забракованную продукцию, прошедшую полный цикл производства;
3) выделенные образцы животной ткани (рассматриваемые как представительная часть медицинского изделия), которые прошли полный цикл процесса производства.
6.4 Для каждой партии медицинских изделий часть раствора жидкого стерилизующего средства или раствора жидкого стерилизующего средства, оставшегося для следующего процесса стерилизации, должна подвергаться проверке на бактерицидную активность в условиях стерилизации партии медицинских изделий с использованием носителя из животной ткани, инокулированного микроорганизмами согласно ГОСТ Р ИСО 11138-1. Носитель должен содержать не менее 10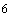 микроорганизмов с известной высокой резистентностью к данному процессу стерилизации, установленной во время проведения аттестации эксплуатируемого оборудования (5.3).
микроорганизмов с известной высокой резистентностью к данному процессу стерилизации, установленной во время проведения аттестации эксплуатируемого оборудования (5.3).
Примечание - При проведении испытания необходимо принять меры предосторожности, чтобы минимизировать риск контаминации производственного оборудования.
6.5 Если основная часть процесса жидкостной химической стерилизации не позволяет вводить тест-микроорганизмы в сосуд для стерилизации или в производственную среду, а также:
a) взаимосвязь между полным химическим составом жидкого стерилизующего средства и его бактерицидной активностью установлена;
b) процесс стерилизации длительно применялся, по крайней мере, для 30 партий медицинских изделий, и при этом не отмечено ни одного отказа согласно 6.4,
то проведение испытаний согласно 6.4 может быть приостановлено, и должен быть проведен полный химический анализ жидкого химического стерилизующего средства, оставшегося после окончания процесса стерилизации, способный подтвердить соответствие заданным предельным значениям характеристик процесса (остаточной бактерицидной активности средства).
Примечание - Объем 30 партий, упомянутый в перечислении b), основан на предположении о биномиальном распределении бракованных изделий при уровне значимости 0,95 (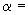 0,05) и доверительном интервале 0,9.
0,05) и доверительном интервале 0,9.
6.6 Результаты, полученные в соответствии с 6.2, 6.3, а также 6.4 или 6.5, должны быть внесены в часть заключения, разрешающего выпуск продукции как стерильной, и должны храниться как часть протоколов стерилизации.
6.7 Все протоколы должны храниться в соответствии с ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002.
7 Выпуск продукции после стерилизации
7.1 Критерии, по которым данный процесс стерилизации признается соответствующим предъявляемым к нему требованиям, должны быть документированы и включать:
a) соответствие технологическим характеристикам процесса;
b) отсутствие роста после инкубации при проведении микробиологического испытания (6.3 и 6.4).
7.2 Процесс стерилизации должен рассматриваться как неудовлетворительный, и продукция должна утилизироваться согласно ГОСТ Р ИСО 9001 или ГОСТ Р ИСО 9002 в следующих случаях:
a) параметр процесса не укладывается в документированные допуски;
b) любое микробиологическое испытание (6.3 и 6.4) показывает рост микроорганизмов после инкубации.
ПРИЛОЖЕНИЕ А
(справочное)
РУКОВОДСТВО
по оценке соответствия настоящему стандарту
А.1 Введение
Настоящее руководство не является контрольным пособием по оценке соответствия настоящему стандарту.
Оно содержит пояснения и методы, пригодные для достижения соответствия установленным требованиям, помогает обеспечить единый подход к пониманию и применению стандарта, при этом могут использоваться также и другие методы, если они являются эффективными в достижении соответствия требованиям стандарта.
Руководство не является исчерпывающим. В руководстве приведены примеры выполнения соответствующих требований. В равной степени возможно использование других методов, дающих такие же результаты. В нем содержатся также рекомендации по выполнению требований стандарта и обращается внимание на требования, не очевидные для людей, детально не знакомых с процессами стерилизации медицинских изделий.
Ссылки на пункты настоящего стандарта приведены в квадратных скобках.
А.2 Персонал [4.2]
Уровень квалификации, подготовки и опыта персонала различных специальностей зависит от выполняемой работы. Общее руководство по подготовке персонала, являющейся частью системы обеспечения качества, приведено в ГОСТ Р ИСО 9004.
Особые требования предъявляются к квалификации и подготовке персонала, отвечающего за:
- микробиологический контроль;
- ветеринарную микробиологию;
- химический анализ и приготовление составов;
- монтаж оборудования;
- обслуживание оборудования;
- механические испытания при аттестации эксплуатируемого оборудования;
- эксплуатацию стерилизатора;
- калибровку;
- разработку процесса;
- технические характеристики оборудования;
- асептическую обработку.
А.3 Разработка процесса и совместимость продукции [4.5]
А.3.1 Разработка процесса стерилизации для конкретного медицинского изделия требует создания такого процесса, который является одновременно эффективным и совместимым с медицинским изделием. Следовательно, исследования совместимости продукта вместе с экспериментальным определением и/или оптимизацией процесса стерилизации могут быть предприняты на стадии разработки продукции.
Требования к системе качества, которые распространяются и на разработку медицинских изделий - по ГОСТ Р ИСО 9001 и ГОСТ Р 51536.
А.3.2 В процессе жидкостной химической стерилизации продукция может подвергаться резким воздействиям окружающей среды, а также вступать в реакцию с используемыми жидкими стерилизующими средствами, поэтому необходимо обеспечить, чтобы функциональное назначение и безопасность продукции не пострадали от воздействия предполагаемых условий стерилизации.
А.3.3. При выборе процесса стерилизации для медицинских изделий необходимо учитывать факторы, влияющие на эффективность процесса (примечание в разделе I):
a) доступность стерилизационного оборудования;
b) диапазон условий, достижимых при применении доступного стерилизационого оборудования;
c) процессы стерилизации, уже используемые для другой продукции;
d) требования к остаточной концентрации химических веществ и/или продуктов их реакции;
e) результаты экспериментов по разработке процесса;
f) возможность того, что использование жидких химических стерилизующих средств приведен к размножению (пролиферации) резистентных микроорганизмов.
А.3.4 Схема разработки процесса стерилизации включает в себя следующие элементы:
a) определение критических параметров процесса и пределов их изменений;
b) оценку бионагрузки продукта, на основе которой должна определяться расчетная микробиологическая контаминация для разработки процесса стерилизации;
c) подтверждение пригодности инокулированного носителя, предназначенного для проведения аттестации эксплуатируемого оборудования и текущего контроля.
В результате мероприятий по разработке процесса стерилизации должны быть определены его характеристики. Пригодность такого стерилизационного процесса должна быть показана при проведении аттестации эксплуатируемого оборудования (5.3).
А.4 Валидация [5]
А.4.1 Оценка бионагрузки при валидации процесса [5.3.3]
Настоящее руководство дополняет руководство, приведенное в ГОСТ Р ИСО 11737-1, поскольку существуют особые трудности в осуществлении оценки бионагрузки животных тканей.
Оценка бионагрузки устанавливает:
- природу контаминирующих микроорганизмов, присутствующих на продукции;
- количество микроорганизмов, присутствующих на продукции;
- пределы колебаний и, следовательно, предсказуемости контаминации сравнением значений, полученных при контроле последовательных партий продукции.
Оценке бионагрузки подлежат:
- исходные материалы животного происхождения;
- материалы после каждой ответственной стадии обработки;
- продукция непосредственно перед стерилизацией (предстерилизационное число).
Любой метод оценки бионагрузки может только показывать присутствие ограниченного числа микроорганизмов и их штаммов. Поэтому значения бионагрузки должны корректироваться с учетом погрешности эффективности извлечения микроорганизмов из продукции и действенности используемых условий культивирования при определении этих микроорганизмов. Эффективность извлечения может колебаться, а также быть крайне низкой для животных тканей. Чтобы компенсировать такую неопределенность, в уравнение для расчета времени выдержки в 5.3.7 вводится коэффициент безопасности, равный 100.
Для обеспечения надежного определения количества выросших колоний необходимо обратить внимание на выбор соответствующих питательных сред и условий культивирования, а также учесть требования к выделению микроорганизмов, которые могут быть связаны с материалами животного происхождения. В некоторых случаях можно использовать среды, приведенные в таблице A.1.
Таблица А.1 - Перечень сведений о питательных средах, условиях инкубации и выращиваемых в них тест-микроорганизмах
|
Питательная среда* |
Возможные условия инкубации*** |
Тест-микроорганизмы для демонстрации ростовых свойств питательных сред**** |
|
1 Триптоно-соевый бульон и триптоно-соевый агар |
Аэробные (30-35 °С) |
Bacillus subtilis |
|
2 Питательный бульон и питательный агар |
Аэробные (30-35 °С) |
Bacillus subtilis |
|
Аэробные (20-25 °С) |
Saccharomyces cerevisiae | |
|
3 Среда Ловенстайна - Дженсена (Левенштейна-Йенсена) |
Аэробные (30-35 °С) |
Trichophyton rubrum |
|
4 Кровяной агар |
Аэробные (30-35 °С) |
Bacillus subtilis |
|
Анаэробные (30-35 °С) |
Clostridium sporogenes | |
|
5 Картофельно-декстрозный агар |
Аэробные (20-25 °С) |
Saccharomyces cerevisiae |
|
6 Среда Робертсона из отварного мяса** |
Аэробные (30-35 °С) |
Clostridium sporogenes |
|
* Среды не должны содержать цветовых индикаторных красителей (которые часто являются ингибиторами микробного роста), за исключением особых случаев, например, среды Ловенстайна-Дженсена Л-Дж. - (Левенштейна-Йенсена). Определение роста в жидких средах должно проводиться визуально, либо по стандарту мутности, либо по плотности, либо путем микроскопирования и в необходимых случаях подтверждаться пересевом на соответствующие твердые питательные среды. | ||
 ).
).
А.4.2 Аттестация в эксплуатации
А.4.2.1 Общие положения
При выборе видов микроорганизмов, используемых при валидации процесса стерилизации материала животного происхождения жидкими химическими стрелизующими средствами, следует рассматривать:
- виды микроорганизмов, которые могут присутствовать в источнике тканей животных;
- виды микроорганизмов, выделенные в время оценки бионагрузки продукции;
- виды микроорганизмов, выделенные из окружающей среды, в которой производится сбор тканей животных, и производственной среды, в которой производится конечное медицинское изделие;
- виды микроорганизмов, которые имеют доказанную высокую резистентность к воздействию жидких химических стерилизующих средств, или которые могут иметь повышенную резистентность;
- вариационный размах видов микроорганизмов.
Примеры микроорганизмов, которые могут быть использованы, приведены в таблице А.2. Перечисленные микроорганизмы ранее показали значительную резистентность к воздействию жидких химических стерилизующих средств на животные ткани. Таблица может рассматриваться только как справочный материал, а не как исчерпывающий перечень микроорганизмов, которые подлежат оценке.
Таблица А.2 - Примеры микроорганизмов, которые использовались для оценки активности некоторых жидких химических стерилизующих средств
|
Вид микроорганизмов |
Номер в коллекции культур | ||
|
ATTC* |
NCTC** |
NCIMB*** | |
|
Споры |
| ||
|
Clostridium sporogenes |
3584 |
- |
10696 |
|
Basillus subtilis |
6051 |
3610 |
3610 |
|
Basillus pumilus |
27142 |
10327 |
10692 |
|
Chaetomium globosum |
6205 |
- |
- |
|
Microascus cinereus |
16594 |
- |
- |
|
Вегетативные клетки |
|
|
|
|
Mycobacterium chelonae |
35752 |
946 |
1474 |
|
Mefhylobacterium extorquens |
43645 |
- |
9399 |
|
Trichosporium beigelii |
22310 |
- |
- |
|
* Американская Коллекция типовых культур. | |||
Три стадии микробиологической аттестации эксплуатируемого оборудования - скрининг, построение кривых выживания и оценка инактивации на носителях из тканей животных - рассмотрены в А.4.2.3-А.4.2.5 соответственно.
А.4.2.2 Нейтрализация [5.3.4]
Перед началом микробиологической аттестации эксплуатируемого оборудования необходимо обеспечить, чтобы на результаты аттестации не повлияли бактерицидные и бактериостатические эффекты от следов жидких химических стерилизующих средств, вносимых в систему выращивания микроорганизмов. Влияние бактерицидных и бактериостатических веществ может быть уменьшено разбавлением, исключено с помощью фильтрации или компенсировано реакцией с нейтрализующим агентом.
Выбор системы нейтрализации будет зависеть от состава жидкого химического стерилизующего средства, а эффективность выбранной системы должна быть показана до начала аттестации.
А.4.2.3 Скрининг (сортирование) микробиологических изолятов [5.3.6 а)]
А.4.2.3.1 Общие положения
Практика показала, что нецелесообразно изучать инактивацию всех изолятов, полученных из продукции, поэтому следует использовать скрининговый тест. Подвергая образцы животных тканей менее жестким условиям воздействия, чем те, которые используются в процессе стерилизации, можно быстро отделить более резистентные изоляты и при изучении инактивации использовать только их. Любой изолят, оказывающийся более резистентным, чем стандартные культуры, выбранные для валидации (таблица А.2), должен быть полностью охарактеризован.
Поскольку применение жидких химических средств в процессах стерилизации связано с индивидуальными особенностями системы, необходимо проявлять постоянное внимание к определению, скринингу и испытанию обнаруживаемых микроорганизмов, которые могут обладать значительной резистентностью к процессу стерилизации. Существует риск того, что во время производства могут быть внесены новые или измененные микроорганизмы, обладающие более высокой резистентностью к процессу стерилизации, чем исходные микроорганизмы, используемые при испытаниях и валидации. Поэтому необходимо установить процедуру скрининга и оценить резистентность микроорганизмов, обнаруженных при производстве и в окружающей среде (процедуру скрининга микробиологических изолятов см. также в А.5).
В процессе микробиологического скрининга обнаружение и оценка новых или измененных микроорганизмов должны осуществляться своевременно.
Процедура скрининга микробиологического изолята должна включать три этапа:
a) сбор микробиологического изолята;
b) идентификацию микробиологического изолята;
c) проведение контрольных испытаний микробиологического изолята на резистентность (скрининг).
А.4.2.3.2 Сбор микробиологического изолята
Микробиологические изоляты должны быть собраны как в производственном процессе, так и из окружающей среды, в которой производится медицинское изделие. Сначала должны быть собраны те микроорганизмы, о которых точно известно, что они присутствуют на продукции до проведения стерилизации (изоляты бионагрузки). В дополнение к бионагрузке продукции изоляты должны быть собраны из производственной среды, в которой изготавливается продукция. Эта среда может включать рабочие растворы, рабочие поверхности, системы очистки воды, сырье и персонал, однако не ограничивается ими.
А.4.2.3.3 Идентификация микробиологического изолята
Микробиологические изоляты, собранные для оценки, должны быть охарактеризованы и/или идентифицированы. Характеристика должна включать, как минимум, морфологию колоний и клеток, окраску по Граму и описание скорости роста. Если возможно, предпочтительно идентифицировать виды или подвиды.
А.4.2.3.4 Проведение испытаний на резистентность (скрининг)
Контрольные испытания микробиологических изолятов на резистентность должны быть проведены в соответствии с 5.3.6. Микробиологические изоляты, проявляющие значительную резистентность к процессу стерилизации при первичном контрольном испытании, должны быть подвергнуты полной оценке и сравнению с микроорганизмами, использованными при первичной аттестации эксплуатируемого оборудования и валидации. Необходимо оценить относительную резистентность контрольных микробиологических изолятов с точки зрения процесса стерилизации.
Один из подходов к первичному контрольному испытанию на резистентность заключается в том, чтобы подвергнуть воздействию жидкого химического стерилизующего средства при минимальных параметрах процесса стерилизации суспензию, содержащую не менее 10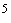 выделенных микроорганизмов, и выдержать ее там в течение времени, равного величине самого резистентного микроорганизма, используемого при аттестации эксплуатируемого оборудования. Если после такой выдержки обнаруживаются жизнеспособные микроорганизмы, то это означает, что резистентность изолята на 20% или более превышает резистентность самого устойчивого микроорганизма, применяемого при аттестации эксплуатируемого оборудования. В этом случае полученный изолят подлежит полной идентификации, а кинематика его инактивации - детальному изучению.
выделенных микроорганизмов, и выдержать ее там в течение времени, равного величине самого резистентного микроорганизма, используемого при аттестации эксплуатируемого оборудования. Если после такой выдержки обнаруживаются жизнеспособные микроорганизмы, то это означает, что резистентность изолята на 20% или более превышает резистентность самого устойчивого микроорганизма, применяемого при аттестации эксплуатируемого оборудования. В этом случае полученный изолят подлежит полной идентификации, а кинематика его инактивации - детальному изучению.
А.4.2.4 Исследование кинетики микробиологической инактивации [5.3.6]
А.4.2.4.1 Общие положения
При проведении аттестации эксплуатируемого оборудования строят кривую инактивации для каждого микроорганизма, причем кривая должна строиться как минимум по пяти точкам, охватывающим, по крайней мере, тысячекратное снижение числа микроорганизмов. Каждая точка обычно строится по трем повторным опытам и должна иметь воспроизводимость в пределах удвоенного стандартного отклонения.
По полученным результатам может быть рассчитана величина наиболее резистентного микроорганизма, используемого в опытах. Расчет величины возможен при условии, что в полулогарифмических координатах (логарифм числа выживших микроорганизмов в зависимости от времени выдержки) кривая выживания является линейной. Если имеются отклонения от линейности, то предсказать параметры удовлетворительного процесса стерилизации трудно. Такие отклонения должны служить предметом дальнейшего изучения в целью лучшей характеристики кинетики инактивации.
Чтобы оценить величину при малых количествах выживших микроорганизмов, можно использовать метод наиболее вероятного числа (НВЧ), проводимый с тремя наиболее резистентными к воздействию данного процесса стерилизации микроорганизмами. Полученная величина может быть использована в качестве критерия приемлемости расчетного времени выдержки.
Определение величины методом НВЧ описано в приложении В к ГОСТ Р ИСО 11135.
А.4.2.4.2 Этапы исследований
Валидацию процессов стерилизации жидкими химическими средствами следует проводить так, чтобы можно было оценить взаимное влияние друг на друга тест-микрорганизмов и медицинского изделия. Это достигается при определении кинетики инактивации тест-микроорганизмов, культивируемых совместно с медицинским изделием или инокулированных на нем.
Такая оценка включает четыре этапа исследований:
- определение компонентов, которые представляют наибольшее сопротивление воздействию жидкого химического стерилизующего средства;
- определение метода, с помощью которого микроорганизмы обнаруживаются на медицинском изделии или на выбранном компоненте;
- валидацию метода восстановления/определения микроорганизмов на медицинском изделии или выбранном компоненте;
- определение кинетики инактивации тест-микроорганизмов в присутствии медицинского изделия или выбранного компонента.
Используют метод прямого подсчета числа выживших микроорганизмов в зависимости от времени обработки (кривая выживания), метод НВЧ (например, его разновидности метод Стамбо-Мэрфи-Кокран (Stumbo-Murphy-Cochran) или метод Спирмен-Карбер (Spearman-Karber)) [9]. Построение кривой выживания предподчтительней, так как этот метод предоставляет достаточные данные для описания кинетики инактивации тест-микроорганизмов в присутствии медицинского изделия или выбранного компонента.
Метод НВЧ может применяться, если медицинское изделие или компонент не позволяют проводить последовательное выявление выжившей популяции микроорганизмов в зависимости от времени обработки (т.е. медицинское изделие или носитель не могут подвергаться вымачиванию для взятия пробы с целью определения популяции выживших микроорганизмов).
Хотя прямой подсчет и построение кривой выживания дают больше информации о кинетике инактивации тест-микроорганизма, они в то же время могут потребовать больше времени и ресурсов. Процедуры прямого подсчета связаны с извлечением всех жизнеспособных микроорганизмов после определенного времени выдержки в жидком химическом стерилизующем средстве. Извлечение может проводиться так же, как при оценке бионагрузки (ГОСТ Р ИСО 11737-1), например, путем гомогенизации испытуемого компонента и приготовления соответствующих разведении для подсчета выживших микроорганизмов.
Гомогенизация испытуемого компонента (например, животных тканей) может быть проведена путем его асептического вымачивания или диспергирования/растворения в стерильном разбавителе. Готовятся последовательные разведения суспензии, и на соответствующих разведениях производится прямой подсчет по стандартным методам (ГОСТ Р ИСО 11737-1).
А.4.2.5 Использование носителей из тканей
А.4.2.5.1 Общие положения
При использовании тест-микроорганизма, показавшего при испытаниях суспензии наибольшую резистентность к воздействию жидкого химического стерилизующего средства, кинетика его инактивации должна изучаться в присутствии испытуемого компонента. Планирование опытов должно включать контролируемые обработки (при худших условиях) инокулированного или культивированного испытуемого компонента в жидком химическом стерилизующем средстве при разных временах выдержки. Образцы должны отбираться через определенные интервалы времени для оценки выжившей популяции валидированным методом восстановления микроорганизмов.
Изучение кинетики инактивации завершается построением кривой выживания в полулогарифмической шкале (на графике в координатах логарифм числа выживших микроорганизмов - время выдержки). Затем рассчитывается величина тест-микроорганизма в присутствии испытуемого компонента и при наихудших условиях стерилизации. Чтобы обеспечить воспроизводимость данных, оценка кинетики инактивации должна проводиться не менее трех раз.
А.4.2.5.2 Выбор носителя
Исследования кинетики микробиологической инактивации должны проводиться на материалах носителя, которые создают наибольшее сопротивление процессу стерилизации. Выбор носителя должен проводиться с учетом обеспечения контакта и/или взаимодействия жидкого химического стерилизующего средства с носителем (например, гидрофобный волокнистый материал может труднее стерилизоваться, чем гладкие гидрофильные поверхности).
Если такой выбор носителя не является очевидным, необходимо провести скрининг для определения наиболее трудностерилизуемых носителей.
А.4.2.5.3 Определение микроорганизма(ов) на испытуемом компоненте (носителе)
Метод определения жизнеспособных микроорганизмов на носителе до начала стерилизации в жидком химическом средстве имеет первостепенную важность при создании условий эффективной стерилизации с точки зрения установления противодействующих стерилизации факторов, встречающихся в практике. Могут быть использованы два метода определения жизнеспособных микроорганизмов на испытываемом компоненте: прямая инокуляция или культивирование при условиях, имитирующих производственные.
При прямой инокуляции используется жизнеспособная споровая или клеточная суспензия, которая наносится на носитель непосредственно перед выдержкой в жидком химическом стерилизующем средстве. Нужно обратить внимание на время, необходимое для проникания инокулята в носитель и связывания его с носителем, прежде чем начать стерилизацию, а также необходимо учесть бактерицидный эффект, который носитель может оказывать на инокулят.
Если выбранный микроорганизм способен давать рост в/на продукции при нормальных производственных условиях, необходимо применять кулитивирование микроорганизмов в условиях, имитирующих производственные, а при возможности делать это и дополнительно к инокуляции. Должен быть установлен метод культивирования носителя(лей) с тест-микроорганизмами. При таких условиях количество жизнеспособных микроорганизмов в/на продукции должно быть не менее 1000 колониеобразующих единиц (КОЕ), В каждой отдельной системе культивирования это количество КОЕ до начала стерилизации должно быть одинаковым в каждом компоненте. Данные для подтверждения соответствующей минимальной популяции и ее однородности должны быть собраны непосредственно перед изучением кинетики инактивации.
Метод восстановления жизнеспособных микроорганизмов носителя после стерилизации жидким химическим средством должен быть определен и валидирован. Валидация должна показать, что выбранный метод обеспечивает воспроизводимое восстановление выживших микроорганизмов.
А.4.2.6 Влияние органических материалов
Для некоторой продукции контаминация органическими материалами может служить фактором, ограничивающим эффективность процесса. Исследования должны включать оценку влияния растворов, содержащих соответствующие органические вещества (например, сыворотки, альбумин и т.д.) или стерильные суспензии тканей. Вид и концентрацию органического материала следует документировать.
Для продукции, которая может высушиваться в присутствии органического материала, должны использоваться инокулированные носители, содержащие микроорганизмы, высушенные в соответствующем органическом материале. Такие инокулированные носители могут включать препараты микроорганизмов, устойчивых к высушиванию, таких как споры Clostridium sporogenes, Bacillus subtilis, Enterococcus faecalis, Mycobacterium chelonae и Candida albicans (таблицы A.1 и A. 2).
A.4.2.7 Валидация асептического процесса [5.3.8]
Асептическое перемещение продукции из стерилизующего вещества в финишный стерильный контейнер также требует валидации. В эту валидацию должны быть включены следующие параметры:
- стерилизация пустых контейнеров в соответствии с требованиями соответствующего стандарта по стерилизации (ГОСТ Р ИСО 11134, ГОСТ Р ИСО 11135 и ГОСТ Р ИСО 11137);
- стерилизация любых консервирующих растворов, в которые асептически переносится продукция;
Примечание - Фильтрующая стерилизация - по ГОСТ Р ИСО 13408-1.
- физический и микробиологический контроль среды, в которой производится асептическое перемещение продукции;
- асептическое перемещение с использованием проб окружающей среды для имитации ее воздействия на перемещаемую продукцию в процесс перемещения из стерилизующего раствора в финишный контейнер.
А.5 Постоянный микробиологический контроль [6.1]
Выделение микроорганизмов и их испытание на резистентность следует проводить с заданной периодичностью. Цель этих испытаний состоит в определении возможного изменения бионагрузки в процессе стерилизации. Поскольку процедуры валидации предназначены для оценки эффективности процесса стерилизации в отношении конкретной группы микроорганизмов, текущее испытание должно показать, что присутствующие в процессе стерилизации микроорганизмы не являются или не стали более резистентными, чем те, которые использовались в начальных валидационных испытаниях (также А.4.2.3.1).
А.6 Управление и контроль процесса [6]
Процесс стерилизации жидким химическим стерилизующим средством обычно включает следующие этапы:
- приготовление жидкого химического стерилизующего средства;
- выдержка продукции в стерилизующем средстве при контролируемой температуре в течение определенного времени.
Если стерилизация не является финишной, то добавляются еще два этапа:
- подготовка и стерилизация первичной упаковки и консервирующего раствора, в которых должна поставляться продукция;
- асептическое перемещение продукции из стерилизующего средства в первичную упаковку.
Приготовление стерилизующего средства требует тщательного контроля. Характеристики составляющих компонентов (номер серии и количество) должны храниться, а конечная концентрация активного ингредиента (ов) должна быть подтверждена экспериментально. Часто стерилизующие растворы фильтруются перед использованием с целью удаления любых микроорганизмов и других загрязнителей, вносимых из компонентов стерилизующего средства. После использования фильтров они должны быть проверены на целостность.
Процесс стерилизации необходимо проводить в сосудах для стерилизации с определенными техническими характеристиками при контролируемых температурных условиях.
Для текущего контроля правильности процесса состав стерилизующего средства должен проверяться после того, как продукция извлечена из раствора. Такие проверки могут быть химическими или микробиологическими. Химический контроль пробы может проводиться с целью подтверждения того, что состав раствора после завершения процесса находится в пределах, установленных технологией. Микробиологическая проверка может представлять собой, например, выдержку инокулированного носителя в стерилизующем растворе для подтверждения остаточной бактерицидной эффективности.
После стерилизации продукция может быть асептически перенесена в свой финишный контейнер. Во время перемещения близлежащая окружающая среда должна подвергаться микробиологическому контролю.
Необходимо утвердить и вести перечень сотрудников, аттестованных на право асептического перемещения продукции. Этот утвержденный список должен постоянно пересматриваться, а персонал периодически подвергаться переаттестации. При аттестации и переаттестации обычно применяют пробное перемещение питательных сред аналогично методу "розлива бульона", используемому при аттестации фильтрующей стерилизации и асептических процессов.
Если продукция выпускается в консервирующем растворе, последний перед использованием должен быть стерилизован. Асептическое производство должно соответствовать требованиям ГОСТ Р ИСО 13408-1.
Испытание конечной продукции имеет ограниченное применение при определении стерильности партии продукции. Однако, в этом частном случае при испытании на микробиологическую контаминацию после стерилизации может быть обнаружен определенный пророст (6.3). При проведении таких микробиологических испытаний конечной продукции важно обеспечить удаление остатков бактерицидного стерилизующего средства или консервирующего раствора (А.4.2.2).
ПРИЛОЖЕНИЕ В
(справочное)
Взаимосвязь российских, международных и европейских стандартов
B.1 Идентичные стандарты
|
ГОСТ Р ИСО |
Стандарт ИСО |
Европейский стандарт |
|
ИСО 9001 |
ЕН 9001 | |
|
ИСО 9002 |
EH 9002 | |
|
- |
ИСО 9004-1 |
EH 9004-1 |
В.2 Технически эквивалентные стандарты
|
ГОСТ Р ИСО |
Стандарт ИСО |
Европейский стандарт |
|
ИСО 11135 |
ЕН 550 | |
|
ГОСТ Р ИСО 11737-1 |
ИСО 11737-1 |
ЕН 1174-1 |
В.3 Сходные стандарты
|
ГОСТ Р ИСО |
Стандарт ИСО |
Европейский стандарт |
|
ГОСТ Р ИСО 11134 |
ИСО 11134 |
ЕН 554 |
|
ИСО 11137 |
ЕН 552 | |
|
ГОСТ Р ИСО 11138-1 |
ИСО 11138-1 |
ЕН 866 |
|
ИСО 13408-1 |
ЕН ХХХХХ* | |
|
ГОСТ Р 51536 |
ИСО 13485 |
ЕН 46001 |
|
ИСО 13488 |
ЕН 46002 | |
|
* Готовится к публикации. | ||
В.4 Не имеющие аналогичных международных стандартов
|
ЕН 556 Стерилизация медицинских изделий. Требования к медицинским изделиям с маркировкой "Стерильно". Sterilization of medical devices. Requirements for medical devices labelled "Sterile" |
|
EH 12442-1* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 1. Анализ и управление риском. Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 1. Analysis and management of risks |
|
EH 12442-2* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 2. Источники, средства контроля, сбор и обращение. Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 2. Sourcing, controls, collection and handling |
|
EH 12442-3* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 3. Удаление и/или инактивация вирусов и других трансмиссивных агентов. Установление нежизнеспособности. Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 3. Elimination and/or inactivation of viruses and other transmissible agents. Determination of non-viability |
|
* Готовится к публикации. |
ПРИЛОЖЕНИЕ С
(справочное)
Библиография
[1] EH 556 Стерилизация медицинских изделий. Требования к медицинским изделиям с маркировкой "Стерильно". EN556 Sterilization of medical devices. Requirements for medical devices tabelled "Sterile"
[2] EH 12442-1* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 1. Анализ и управление риском. EN 12442-1 Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 1. Analysis and management of risks
[3] EH 12442-2* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 2. Источники, средства контроля, сбор и обращение. EN 12442-2 Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 2. Sourcing, controls, collection and hahdiing
[4] EH 12442-3* Животные ткани и их производные, используемые при производстве медицинских изделий. Часть 3. Удаление и/или инактивация вирусов и других траснмиссивных агентов. Установление нежизнеспособности. EN 12442-3 Animal tissues and their derivatives utilized in the manufacture of medical devices. Part 3. Elimination and/or inactivation of viruses and other transmissible agents. Determination of non-viability
[5] Комиссия Европейских Сообществ (1989). Руководящие правила для лекарственных препаратов в Европейском Сообществе, т. IV, Руководство по GMP для лекарственных препаратов - Commission of the European Communities (1989). The Rules Governing Medicinal Products in the European Community, Vol.IV, Guide to good manufacturing practice for medicinal products
[6] Руководящие указания по минимизации риска передачи агентов, вызывающих губчатые энцефалопатии, через лекарственные препараты. - Commission of the European Communities (1992). Committee Medicinal Products; Ad Hoc Working Party on Biotechnology/Pharmacy - Guidelines for minimizing the risk of transmission of agents causing spongiform encephalopaties via medicinal products (11113298191- EN)
[7] Директива Совета 64/433/EEC от 26 июня 1964 г. по проблемам торговли свежим мясом внутри сообщества - Directive 64/433/EEC, Council Directive of 26 June 1964 on health problems affecting intra-community trade in fresh meat (OJ 1121 29107164)
[8] Директива Совета 93/42/ЕЕС от 26 июня 1993 г. по медицинским изделиям - Directive 93/42/ЕЕС, Council Directive of 14 June 1993 on Medical devices
[9] Пфлюг И.Ю. и Холкомб Р.Г. Принципы термической деструкции микроорганизмов - Pflug I.J and Holcomb R.G., Principles of Thermal Destruction of Microorganisms, In Disinfection Sterilization and Preservation. 3 rd edition. S.S. Block, ed, Philadelphia (PA): Lea and Febiger, 1983
__________________
* Готовится к публикации.
Текст документа сверен по:
официальное издание
М.: ИПК Издательство стандартов, 2003