
- USD ЦБ 03.12 30.8099 -0.0387
- EUR ЦБ 03.12 41.4824 -0.0244
|
Краснодар:
|
погода |
01
:
56
апреля
15
вторник,
Курсы
Индексы
- DJIA 03.12 12019.4 -0.01
- NASD 03.12 2626.93 0.03
- RTS 03.12 1545.57 -0.07
ГОСТ Р 52458-2005
(ИСО 11979-7:2001)
Группа П46
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Имплантаты офтальмологические
ИНТРАОКУЛЯРНЫЕ ЛИНЗЫ
Часть 7
Клинические испытания
Ophthalmic implants. Intraocular lenses.
Part 7.
Clinical investigations
ОКС 11.040.40
ОКП 94 8100
Дата введения 2007-01-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Открытым акционерным обществом "ТКС-оптика" на основе собственного аутентичного перевода стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 296 "Оптика и оптические приборы" и подкомитетом ПК 7 "Офтальмологическая оптика"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 28 декабря 2005 г. N 374-ст
4 Настоящий стандарт является модифицированным по отношению к международному стандарту ИСО 11979-7:2001 "Имплантаты офтальмологические. Интраокулярные линзы. Часть 7. Клинические испытания" (ISO 11979-7:2001 "Ophthalmic implants - Intraocular lenses - Part 7: Clinical investigations") путем:
- изменения его структуры. Сопоставление структуры настоящего стандарта со структурой указанного международного стандарта приведено в дополнительном приложении G;
- введения дополнительных слов (фраз). При этом дополнительные слова (фразы), включенные в текст стандарта для учета потребностей национальной экономики Российской Федерации и/или особенностей национальной стандартизации, выделены курсивом;
- введения приложения В, которое дополняет разделы и приложения указанного международного стандарта с учетом потребностей национальной экономики Российской Федерации и/или особенностей национальной стандартизации;
- исключения приложения А примененного международного стандарта, которое нецелесообразно применять в национальной стандартизации в связи с тем, что оно является справочным и содержит термины, приведенные в ГОСТ Р 51892-2002 (ИСО 11979-1:99).
При ссылке на ИСО 14155-1:2003 "Клинические испытания медицинских изделий. Часть 1. Общие требования" (ISO 14155-1:2003 "Clinical investigation of medical device for human subjects - Part 1: General requirements") в настоящем стандарте дополнительно приведены содержание соответствующих пунктов и приложение Е данного международного стандарта, которые выделены вертикальной линией, расположенной слева от текста. Это обусловлено тем, что международный стандарт не имеет аналогов среди национальных стандартов
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
1 Область применения
Настоящий стандарт распространяется на переднекамерные и заднекамерные интраокулярные линзы (далее - ИОЛ) независимо от материала, из которого они изготовлены, места локализации в глазу пациента и функционального назначения (протез хрусталика глаза или линза, предназначенная для коррекции афакии и аномалий рефракции).
Стандарт устанавливает методы клинических испытаний модификаций ИОЛ, а также требования к протоколам клинических испытаний ИОЛ.
Примечание - Другие типы ИОЛ подвергают клиническим испытаниям по [1].
2 Нормативные ссылки
В настоящем стандарте использованы ссылки на следующие стандарты:
ГОСТ Р 51892-2002 (ИСО 11979-1-99) Имплантаты офтальмологические. Интраокулярные линзы. Часть 1. Термины и определения
ГОСТ Р 52038-2003 (ИСО 11979-2-99) Имплантаты офтальмологические. Интраокулярные линзы. Часть 2. Оптические свойства и методы испытаний
ГОСТ Р 52039-2003 (ИСО 11979-3-99) Имплантаты офтальмологические. Интраокулярные линзы. Часть 3. Механические свойства и методы испытаний
ГОСТ Р 52040-2003 (ИСО 11979-6-99) Имплантаты офтальмологические. Интраокулярные линзы. Часть 6. Срок годности и стабильность при транспортировании
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии сети Интернет или по ежегодно издаваемому информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по соответствующим ежемесячно издаваемым информационным указателям, опубликованным в текущем году. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться замененным (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Термины и определения
3.1 В настоящем стандарте применены термины по ГОСТ Р 51892, ГОСТ Р 52039 и следующие термины с соответствующими определениями:
3.1.1 серьезный неблагоприятный исход (adverse device effect): Послеоперационный исход, потенциально угрожающий зрению.
Примечание - Примеры серьезных неблагоприятных исходов приведены в таблицах А.1 и А.2 приложения А.
3.1.2 план клинических испытаний; ПКИ (clinical investigation plan): Документ, определяющий цели, задачи, методологию, систему контроля, проведения анализа и хранения данных о клинических испытаниях.
3.1.3 клиническое испытание (clinical investigation): Любое разработанное и запланированное систематическое изучение с целью проверки безопасности и/или эксплуатационных свойств ИОЛ с участием субъектов.
3.1.4 субъект (subject): Лицо, участвующее в клинических испытаниях с применением испытуемой ИОЛ и/или в составе контрольной группы.
3.1.5 клинический испытатель (clinical investigator): Лицо и/или организация, ответственные за проведение клинических испытаний и несущие клиническую ответственность за благополучие вовлеченных субъектов.
Примечание - Лицо и/или организация назначаются в соответствии с действующим законодательством.
3.1.6 организатор (sponsor): Лицо и/или организация, которые несут ответственность за начало и/или проведение клинических испытаний.
3.1.7 главный клинический испытатель (principal clinical investigator): Клинический испытатель, ответственный за организацию клинических испытаний, проводимых в одной организации (лаборатории).
3.1.8 координатор (coordinating clinical investigator): Клинический испытатель, назначаемый организатором для координации клинических испытаний, проводимых в нескольких организациях (лабораториях).
3.1.9 клиническая эффективность (clinical performance): Изучение определенного типа ИОЛ и/или его потребительских свойств в случае конкретного использования на субъектах, для которых оно предназначено.
3.1.10 форма отчета (case report form): Документ, предназначенный для записи всей информации, предоставляемой организатору по каждому субъекту в соответствии с требованиями плана клинических испытаний.
3.1.11 журнал клинического испытателя (clinical investigator's brochure): Документ, предназначенный для записи клинической и неклинической информации об испытуемой ИОЛ, относящейся к испытаниям на субъектах.
3.1.12 наблюдатель (monitor): Лицо, назначенное организатором для наблюдения за соответствием действий клинического испытателя протоколу клинических испытаний и проверки исходных данных.
3.1.13 исходные данные (source documents): Информация, представленная в виде оригинальных, идентифицированных и сертифицированных записей клинических осмотров, наблюдений и/или других действий в течение клинических испытаний, необходимая для оценки клинических испытаний.
3.1.14 окончательный отчет (final report): Документ, заполняемый после окончания клинических испытаний, содержащий описание и оценку результатов.
3.1.15 комитет по этике (ethics committee): Независимый компетентный орган, отвечающий за безопасность, благополучие и права субъектов, принимающих участие в клинических испытаниях.
3.1.16 расписка об информированности (informed consent): Документ, имеющий юридическую силу, подтверждающий согласие субъекта на добровольное участие в клинических испытаниях.
Примечание - Требования к расписке об информированности приведены в приложении В.
3.1.17 контрольная группа (control populations): Субъекты, которым имплантированы ИОЛ, соответствующие требованиям ГОСТ Р 52038 - ГОСТ Р 52040.
3.1.18 историческая контрольная группа (historical control population): Контрольная группа, прошедшая клинические испытания и имеющая серьезные неблагоприятные исходы.
4 Этические соглашения
4.1 Общие требования
|
4.1.1 Права, безопасность и добровольность участия субъектов клинических испытаний должны быть защищены в соответствии с этическими принципами, изложенными в Хельсинской декларации, и должны соблюдаться на любом этапе клинических испытаний. |
5 Требования
5.1 Общие требования
|
5.1.1 Между организатором, испытателем и другими лицами, участвующими в клинических испытаниях, должны существовать формальные соглашения, определяющие круг их обязанностей. Все формальные соглашения должны быть задокументированы в письменном виде и подписаны всеми участниками.
5.1.2 Все лица, участвующие в проведении клинических испытаний, должны иметь соответствующую квалификацию/образование и/или опыт для исполнения своих обязанностей. |
5.2 Дополнительные требования
5.2.1 Организатор и клинические испытатели должны оценивать уровни неблагоприятных исходов и остроты зрения для ИОЛ при клинических испытаниях.
5.2.2 Безопасность и эффективность каждой модели ИОЛ должны быть подтверждены:
- клиническими испытаниями, проведенными в соответствии с требованиями настоящего стандарта;
- сравнением параметров модели с параметрами базовой модели, показывающим, что модель является незначительной модификацией базовой модели, безопасность и эффективность которой доказаны клиническими испытаниями.
Примечание - Руководство по определению модификации базовой модели ИОЛ приведено в приложении С.
5.2.3 Клинические испытания ИОЛ, предназначенных для коррекции афакии у взрослых субъектов, должны проводиться организатором с использованием элементов протоколирования по приложению D. Организатор должен разработать протокол с учетом оценки различий постоянных неблагоприятных исходов у испытуемых субъектов и субъектов из контрольной группы.
Если ИОЛ предназначена для расположения в передней и задней камерах глаз, то должны быть проведены отдельные клинические испытания для проверки клинической эффективности ИОЛ в каждой глазной камере.
5.2.4 Все субъекты клинических испытаний в период испытаний должны проходить периодические обследования. Клинические испытания считают оконченными, когда на все субъекты, вовлеченные в испытания, включая тех, у которых ИОЛ были извлечены или заменены, будет составлен окончательный отчет по приложению Е.
6 Методология
6.1 Требования к документации
|
6.1.1 Журнал клинического испытателя и документы по 6.1.3 должны быть подготовлены до начала клинических испытаний. Предоставляемая клиническим испытателем информация должна быть задокументирована. |
Примечание - Обзор научной литературы включает в себя получение информации об оптических и механических свойствах ИОЛ;
|
б) общее описание ИОЛ; |
6.2 Требования к доступу информации
|
6.2.1 Каждый клинический испытатель, участвующий в клинических испытаниях, должен иметь право доступа к необходимой предклинической информации. Вся полученная информация должна храниться конфиденциально. |
6.3 Требования к дополнительному медицинскому обслуживанию
|
6.3.1 Требования к медицинскому обслуживанию приведены в 4.1.3. |
6.4 Требования к плану клинических испытаний
6.4.1 Общие требования
Требования к плану клинических испытаний приведены в [2] (подраздел 4.2).
6.4.2 Дополнительные требования
6.4.2.1 Испытуемая линза должна быть имплантирована только в один глаз субъекта.
6.4.2.2 Клиническим испытаниям в каждый отчетный период подвергают минимальное число образцов ИОЛ. Организатор должен убедиться в наличии необходимого числа субъектов для обеспечения клинических испытаний необходимым числом образцов ИОЛ.
Примечание - Примеры клинических предоперационных/операционных и послеоперационных форм отчета приведены в таблицах D.1-D.5 приложения D.
6.5 Требования к организатору клинических испытаний
|
6.5.1 Перед началом клинических испытаний организатор должен определить, установить, обеспечить и распределить все связанные с испытаниями обязанности и функции. | |
|
1) всех документов по 6.1.3, | |
|
п) обеспечение системы учета применяемых и неиспользуемых ИОЛ. | |
6.6 Требования к наблюдателю
|
6.6.1 Наблюдатель является ответственным лицом за: |
6.7 Требования к клиническому испытателю
6.7.1 Общие требования
|
6.7.1.1 Если клиническим испытателем является организация, то в качестве ее представителя должно быть назначено лицо, обладающее необходимой квалификацией и выполняющее обязанности, указанные в 6.7.1.2-6.7.1.3. |
6.7.2 Дополнительные требования
6.7.2.1 Клинический испытатель должен немедленно сообщать организатору о всех случаях серьезного неблагоприятного исхода и указывать в отчетах о любых других неблагоприятных исходах.
7 Требования к представлению результатов клинических испытаний
|
7.1 Окончательный отчет о клинических испытаниях представляют даже в том случае, если клинические испытания прекращены преждевременно. |
Приложение А
(справочное)
Примеры серьезных неблагоприятных исходов
для переднекамерных и заднекамерных ИОЛ и показателей остроты зрения
А.1 Примеры серьезных неблагоприятных исходов для переднекамерных и заднекамерных ИОЛ приведены в таблицах А.1 и А.2.
Таблица А.1 - Соотношения неблагоприятных исходов для переднекамерных ИОЛ
|
Неблагоприятный исход |
Контрольное отношение, % |
Число субъектов 100 |
Число субъектов 300 | ||
|
Пороговое отношение, % |
Разрешенное число |
Пороговое отношение, % |
Разрешенное число | ||
|
Кумулятивный: |
|||||
|
цистоидный макулярный отек |
10,0 |
19,5 |
15 |
15,0 |
39 |
|
гифема |
4,3 |
11,2 |
8 |
7,7 |
19 |
|
гипопион |
0,2 |
2,9 |
1 |
1,4 |
2 |
|
внутриглазная инфекция |
0,2 |
2,9 |
1 |
1,4 |
2 |
|
смещение линзы из передней камеры |
1,1 |
5,4 |
3 |
3,2 |
6 |
|
блокировка зрачка |
2,0 |
7,8 |
5 |
4,5 |
10 |
|
отслойка сетчатки |
1,2 |
5,4 |
3 |
3,4 |
7 |
|
повторное хирургическое вмешательство (исключая отслойку сетчатки и заднюю капсулотомию) |
2,6 |
8,5 |
5 |
5,2 |
13 |
|
стромальный отек роговицы |
0,5 |
4,2 |
2 |
2,2 |
4 |
|
цистоидный макулярный отек |
3,3 |
10,5 |
7 |
7,1 |
17 |
|
раздражение |
0,9 |
5,4 |
3 |
3,0 |
6 |
|
повышенное внутриглазное давление, требующее лечения |
2,1 |
7,8 |
5 |
4,5 |
11 |
|
Примечания | |||||
Таблица А.2 - Соотношения неблагоприятных исходов для заднекамерных ИОЛ
|
Неблагоприятный исход |
Контрольное отношение, % |
Число субъектов 100 |
Число субъектов 300 | ||
|
Пороговое отношение, % |
Разрешенное число |
Пороговое отношение, % |
Разрешенное число | ||
|
Кумулятивный: |
|||||
|
цистоидный макулярный отек |
3,0 |
9,2 |
6 |
5,9 |
14 |
|
гифема |
2,2 |
7,8 |
5 |
4,5 |
11 |
|
гипопион |
0,3 |
2,9 |
1 |
1,8 |
3 |
|
внутриглазная инфекция |
0,1 |
2,9 |
1 |
1,0 |
1 |
|
смещение линзы из передней камеры |
0,1 |
2,9 |
1 |
1,0 |
1 |
|
блокировка зрачка |
0,1 |
2,9 |
1 |
1,0 |
1 |
|
отслойка сетчатки |
0,3 |
2,9 |
1 |
2,9 |
3 |
|
повторное хирургическое вмешательство (исключая отслойку сетчатки и заднюю капсулотомию) Повторяющийся: |
0,8 |
4,2 |
2 |
2,6 |
5 |
|
стромальный отек роговицы |
0,3 |
2,9 |
1 |
1,8 |
3 |
|
цистоидный макулярный отек |
0,5 |
4,2 |
2 |
2,2 |
4 |
|
раздражение |
0,3 |
2,9 |
1 |
1,8 |
3 |
|
повышенное внутриглазное давление, требующее лечения |
0,4 |
4,2 |
2 |
1,8 |
3 |
|
Примечания | |||||
А.2 Примеры показателей послеоперационной остроты зрения приведены в таблицах А.3 и А.4.
Таблица А.3 - Средние показатели послеоперационной остроты зрения 0,5 (6/12, 20/40) отн. ед. и/или выше
|
Тип ИОЛ |
Контрольное отношение, % |
Число субъектов 100 |
Число субъектов 300 | ||
|
Пороговое отношение, % |
Требуемое число, не менее |
Пороговое отношение, |
Требуемое число, не менее | ||
|
Переднекамерная ИОЛ |
80,4 |
69,6 |
74 |
74,3 |
230 |
|
Заднекамерная ИОЛ |
92,5 |
84,4 |
88 |
88,2 |
270 |
|
Примечания | |||||
Таблица А.4 - Лучшие показатели послеоперационной остроты зрения 0,5 (6/12, 20/40) отн. ед. и/или выше
|
Тип ИОЛ |
Контрольное отношение, % |
Число субъектов 100 |
Число субъектов 300 | ||
|
Пороговое отношение, % |
Требуемое число, не менее |
Пороговое отношение, |
Требуемое число, не менее | ||
|
Переднекамерная ИОЛ |
90,1 |
81,1 |
85 |
85,4 |
262 |
|
Заднекамерная ИОЛ |
96,7 |
91,0 |
94 |
93,6 |
285 |
А.3 Приведенные в таблицах А.1 и А.2 исследуемые отношения для 100 и 300 субъектов для клинических испытаний будут меньше приведенных пороговых отношений, так как любое статистическое сравнение содержит в себе ошибку выборки. Аналогично исследуемые отношения в таблицах А.3 и А.4 будут больше, чем приведенные пороговые отношения. В интервал между контрольным и пороговым отношениями, приведенными в таблицах, попадают 80% возможных отношений. При этом, чем ближе пороговое отношение к контрольному, тем меньше 80% возможных отношений туда попадут, т.е. возникает ошибка второго типа.
А.4 Приведенные в таблицах А.1 и А.2 расчетные результаты неблагоприятных исходов были получены в результате использования биноминального распределения по формуле
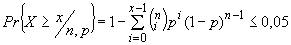 , (А.1)
, (А.1)
где 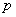 - контрольное отношение (т.е. отношение неблагоприятных исходов для контрольной группы);
- контрольное отношение (т.е. отношение неблагоприятных исходов для контрольной группы);
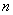 - размер выборки;
- размер выборки;
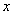 - число зарегистрированных во время клинического испытания неблагоприятных исходов;
- число зарегистрированных во время клинического испытания неблагоприятных исходов;
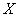 - число неблагоприятных исходов.
- число неблагоприятных исходов.
А.5 Биноминальное распределение используют для того, чтобы проверить, является ли исследуемое отношение неблагоприятных исходов меньше или равным отношению для исторической контрольной группы. Аналогично для таблиц А.3 и А.4 биноминальное распределение используют для того, чтобы проверить, является ли исследуемое отношение остроты зрения 0,5 отн. ед. и выше более или равным отношению для контрольной группы. Альтернативная гипотеза состоит в том, что исследуемое отношение менее отношения для контрольной группы.
А.6 Максимальное значение благоприятных исходов (таблица А.4) может быть получено с использованием биноминального распределения по формуле
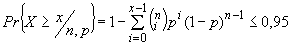 , (А.2)
, (А.2)
где - контрольное отношение для остроты зрения 0,5 отн. ед. и выше;
- размер выборки;
- число зарегистрированных успешных исходов испытания;
- число неблагоприятных исходов.
Приложение В
(рекомендуемое)
Требования к расписке об информированности
В.1 Общие требования
Расписка об информированности должна быть получена от субъекта в письменном виде перед началом участия субъекта в клинических испытаниях.
Примечание - Форма расписки обычно состоит из информационной части и согласительной/подписной части. Эти две части могут быть либо скомбинированы в одном документе (информация для пациента и форма расписки) или разделены на лист информации для пациента и форму расписки пациента.
В.2 Процесс получения расписки об информированности
При получении расписки об информированности следует соблюдать следующие правила:
а) избегать любого принуждения или незаконного влияния на субъект для обеспечения его участия;
б) не уклоняться или не пытаться уклониться от соблюдения законных прав субъекта;
в) использовать язык, который не является специальным и понятен субъекту или его законному представителю;
г) предоставить субъекту время для обдумывания решения об участии;
д) расписка должна содержать датированные подписи субъекта или его законного представителя и клинического испытателя;
е) включать субъект в план клинических испытаний.
В.3 Информация, предоставляемая субъекту для получения расписки об информированности
На понятном для субъекта или его законного представителя языке должна быть изложена следующая информация:
а) описание испытания/назначение:
1) исследования, включенные в испытания,
2) цель испытаний,
3) предполагаемая длительность испытаний и степень вовлеченности субъекта в процесс испытаний,
4) описание исследуемой ИОЛ,
5) описание процедур, исследований с акцентированием внимания субъекта на эксперименте;
б) предсказуемые риски:
1) информация о любых предсказуемых рисках и осложнениях,
2) информация о любых возможных побочных эффектах;
в) потенциальные выгоды:
1) описание потенциальных выгод для субъекта,
2) описание потенциальных выгод для других субъектов;
г) альтернативная терапия:
1) информация об альтернативных методах лечения или допустимых процедурах;
д) конфиденциальность:
1) указание о том, что участие субъекта является конфиденциальным,
2) указание о том, что субъект должен разрешить доступ к своим медицинским записям полномочным лицам и представителям организатора,
3) информация о том, что результаты испытаний могут быть опубликованы без раскрытия личности субъекта;
е) компенсация (медицинская/финансовая):
1) информация об условиях для получения компенсации в случае ранения, возникшего в результате участия в клинических испытаниях, и о том, какие дополнительные меры по охране здоровья будут предоставлены субъекту в случае серьезного увечья,
2) информация о финансовой компенсации за участие, если это предусмотрено;
ж) вопросы о начале и/или окончании клинических испытаний:
1) информация о том, кто должен ответить на вопросы субъекта по исследованиям,
2) информация о том, кто должен ответить на вопросы субъекта в случае травмы,
3) описание обстоятельств, при которых участие субъекта в испытаниях может быть прекращено испытателем;
и) новые открытия:
1) информация о том, что новые открытия, сделанные с участием субъекта, должны быть доступны всем клиническим испытателям.
В.4 Заявления в расписке об информированности
Информационная расписка должна содержать:
а) заявление о том, что участие субъекта является добровольным;
б) заявление о том, что отказ от участия не несет никаких штрафных санкций по отношению к субъекту;
в) заявление о том, что отказ от участия в любой момент клинических испытаний не несет никаких штрафных санкций по отношению к субъекту;
г) заявление о возможных последствиях испытаний;
д) заявление о понимании представленной информации.
В.5 Соглашения в расписке об информированности
При подписании расписки об информированности субъект или его официальный представитель должен быть согласен:
а) на участие в клинических испытаниях;
б) что его персональный врач будет информирован о его участии; или подписать заявление об отказе в представлении такой информации;
в) на использование его персональных данных для целей клинического испытания.
Приложение С
(рекомендуемое)
Руководство по определению модификации базовой модели ИОЛ
С.1 Модификация конструкции/материала базовой модели ИОЛ
С.1.1 Общие положения
Требования к испытаниям ИОЛ, являющимся модификациями базовой модели ИОЛ, уже прошедшей клинические испытания, зависят от степени изменений, внесенных в модифицированную модель ИОЛ. Модификации базовой модели ИОЛ подразделяют на два уровня: уровень А и уровень В. Примеры модификации уровня А и уровня В базовой модели ИОЛ приведены в С.2.
С.1.2 Уровень А
С.1.2.1 Модификации базовой модели ИОЛ уровня А в большинстве случаев не требуют проведения клинических испытаний. В случае, если модифицированная модель ИОЛ обладает свойствами, которые не могут быть адекватно исследованы в период предклинических испытаний, например, потенциальное повреждение тканей вследствие изменения техники имплантации ИОЛ, клинические испытания должны проводиться.
С.1.2.2 Модификацию базовой модели ИОЛ уровня А обычно не используют в качестве базовой модели для последующих модификаций.
С.1.2.3 В случае, если модель ИОЛ находится в стадии клинических испытаний, модификация уровня А этой модели также может быть подвергнута клиническим испытаниям и данные испытаний базовой модели и модификации уровня А могут быть скомбинированы. Если изменения базовой модели ИОЛ уровня А включают замену материала базовых моделей, например, модификации, описанные в С.2.2.12, такую модель не допускается использовать для проведения клинических испытаний.
С.1.3 Уровень В
С.1.3.1 Клинические испытания ИОЛ, являющихся модификациями базовой модели уровня В, должны включать не менее 100 субъектов, наблюдаемых в послеоперационный период по форме отчета в соответствии с таблицей D.4 приложения D.
Число отчетов для всех посещений субъектов должно быть не менее 100.
С.1.3.2 Число ИОЛ, необходимых для проведения клинических испытаний базовой модели ИОЛ уровня В, должно быть минимальным, но достаточным для сравнения отношений неблагоприятных исходов и изменения остроты зрения для испытуемой ИОЛ с отношениями для исторической контрольной группы. Если организатор сравнивает качество испытуемых ИОЛ с одновременно запущенной контрольной группой, то он должен отобрать такое число субъектов, которое будет достаточным для выявления изменений остроты зрения и неблагоприятных исходов с точностью, эквивалентной сравнению с исторической контрольной группой.
С.1.3.3 Число ИОЛ модификации уровня В, не выдержавших испытаний, должно быть не более 10%. Каждый испытатель должен подвергать испытаниям не менее 20% и не более 25% общего числа испытуемых субъектов. Клинические испытания ИОЛ модификации уровня В считают завершенными, когда на все вовлеченные субъекты составлены отчеты в соответствии с таблицей D.4 приложения D.
С.1.3.4 Модификацию ИОЛ уровня В считают базовой моделью, если получены отчеты по не менее чем 100 субъектам (однако не в комбинации с отчетами модификации уровня А в период клинических испытаний), и в случае, если модификация ИОЛ соответствует всем требованиям ГОСТ Р 52038, ГОСТ Р 52039, ГОСТ Р 52040.
С.2 Примеры модификаций ИОЛ
С.2.1 Общие положения
С.2.1.1 Модификации ИОЛ, подвергаемых клиническим испытаниям, подразделяют на модификации базовой модели уровня А и уровня В. Критерий деления приведен в С.2.2 и С.2.3.
Допускается модифицировать базовые модели ИОЛ, имеющие следующие признаки:
- Р - заднекамерные ИОЛ из полиметилметакрилата (ПММА);
- А - переднекамерные ИОЛ из ПММА;
- SP - заднекамерные ИОЛ из мягких материалов, имеющие плоский дизайн (без петель);
- SS - составные заднекамерные ИОЛ с оптической частью, изготовленной из мягких материалов и петлями из стандартных материалов (ПММА, полипропилен или полиамид);
- SN - составные заднекамерные ИОЛ с оптической частью из мягких материалов и петлями из нестандартных материалов.
С.2.1.2 Модифицированная модель может включать различные комбинации модификаций, указанных в С.2.2-С.2.3, если соблюдаются все критерии (например, модифицированная модель может иметь большую оптическую часть со слегка измененной конфигурацией петель и большим общим диаметром). Модификация, указанная в С.2.2.12, отличается от других модификаций тем, что заменен материал или изменена конструкция базовой модели.
С.2.2 Модификации уровня А
С.2.2.1 Зеркально отраженная версия модели ИОЛ
Признаки модифицированных ИОЛ типов P/A/SP/SS/SN должны соответствовать указанным в С.2.1.1.
С.2.2.2 Изменение общего диаметра
Признаки модифицированных ИОЛ для пациентов с определенным диапазоном ширины передней камеры глаза для модифицированных ИОЛ уровня А должны соответствовать указанным в С.2.1.1.
С.2.2.3 Изменения конструкции петель
Признаки модифицированных ИОЛ в случае добавления закруглений или маленьких петель для типов P/A/SS/SN должны соответствовать указанным в С.2.1.1.
С.2.2.4 Изменения наклона петель
Признаки модифицированных ИОЛ в случае изменения плоского дизайна на дизайн с телом, отклоненным назад от петель, приводящий к увеличению саггитали не более чем на 1,6 мм для ИОЛ с задней вершинной рефракцией 20 Дптр, для типов P/SS/SN должны соответствовать указанным в С.2.1.1.
С.2.2.5 Изменения конфигурации, толщины или ширины петель (калибра)
С.2.2.5.1 Общие положения
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Для того, чтобы определить модификацию уровня А, необходимо провести анализ изменения механических свойств ИОЛ.
Если модифицированная модель ИОЛ обладает свойствами, которые не могут быть адекватно исследованы в период предклинических испытаний, например, если может произойти потенциальное повреждение тканей вследствие изменения в технике имплантации, то такая модификация не может быть признана модификацией ИОЛ уровня А.
Методы сравнения механических свойств базовой модели и модифицированной модели приведены в С.2.2.5.2. Если организатор приходит к выводу о том, что по одному методу сравнения модификация не относится к уровню А, он должен указать другой метод до начала клинических испытаний.
Примечание - Методы и анализ данных, применяемых в испытаниях механических свойств, - по ГОСТ Р 52039 (В.2.2.5.2-В.2.2.5.3).
С.2.2.5.2 Сравнение с единичной базовой моделью
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Для сравнения модифицированной модели и модели, подвергаемой клиническим испытаниям, организатор должен доказать, что механические свойства модифицированной и испытуемой моделей аналогичны.
Сравнивая модифицированную и базовую модели ИОЛ, организатор должен доказать, что механические свойства модифицированной ИОЛ аналогичны механическим свойствам базовой модели.
С.2.2.5.3 Сравнение нескольких базовых моделей
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Сравнивая модифицированную и несколько базовых моделей ИОЛ, организатор должен доказать, что механические свойства модифицированной ИОЛ соответствуют характеристикам базовых моделей.
С.2.2.6 Изменение диапазона оптической силы
Признаки модифицированных ИОЛ типов P/A/SP/SS/SN должны соответствовать указанным в С.2.1.1.
Организатор может проводить любые изменения диапазона оптической силы, если ИОЛ соответствует требованиям ГОСТ Р 52038.
Организатор должен помнить о том, что при определенных комбинациях материала и профиля некоторые модели ИОЛ не соответствуют требованиям ГОСТ Р 52038 вследствие влияния сферических аберраций.
С.2.2.7 Изменения размеров оптической части и/или конструкции тела ИОЛ
Признаки модифицированных ИОЛ типов P/SP/SS/SN должны соответствовать указанным в С.2.1.1.
Изменения конструкции тела и/или размеров оптической части допускаются при условии, если общая длина ИОЛ не менее 5,0 мм и не более 7,5 мм вдоль любого меридиана.
С.2.2.8 Добавление, удаление или смещение позиционных отверстий
Признаки модифицированных ИОЛ типов P/A/SP/SS/SN должны соответствовать указанным в С.2.1.1.
Позиционные отверстия, влияющие на качество оптической части, должны располагаться на расстоянии не менее 2,25 мм от центра оптической части для того, чтобы рассеяние и/или искажения от них были минимальными.
С.2.2.9 Добавление рубца на задней поверхности тела ИОЛ
Признаки модифицированных ИОЛ типов Р должны соответствовать указанным в С.2.1.1.
Допускается добавлять рубец на заднюю поверхность тела ИОЛ в случае, если дизайн рубца подтвержден клиническими испытаниями модели ИОЛ с рубцом и край рубца находится на расстоянии не менее 2,25 мм от центра оптической части.
С.2.2.10 Испытание различных конструкций ИОЛ в одном клиническом испытании
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.
Один материал ИОЛ может быть испытан для более чем одной конструкции ИОЛ, если конструкции ИОЛ подтверждены на соответствие базовым моделям. При этом механические свойства испытуемой ИОЛ должны быть аналогичны механическим свойствам базовой модели.
С.2.2.11 Испытание различных материалов в одном клиническом испытании
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.
Одна конструкция ИОЛ может быть испытана с более чем одним материалом для петель, если все материалы до испытаний были подтверждены на соответствие базовым моделям. При использовании более одного материала для петель, все модели ИОЛ должны быть модификациями уровня А и соответствовать требованиям С.2.2.5.
С.2.2.12 Замена материала и конструкции ИОЛ
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Материалы и конструкции базовых моделей могут быть заменены, если использование материала новой (для данного материала) конструкции ИОЛ не приводит к изменению механических свойств базовой модели ИОЛ.
С.2.3 Модификации уровня В
С.2.3.1 Изменение общего диаметра
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Изменение общего диаметра ИОЛ может привести к созданию модификации ИОЛ, не соответствующей требованиям С.2.2.5 при ее сравнении с базовой моделью или с рядом характеристик нескольких базовых моделей.
С.2.3.2 Изменения конфигурации петель
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Изменение конфигурации петель может привести к созданию модификации ИОЛ, не соответствующей требованиям С.2.2.5 при ее сравнении с базовой моделью или с рядом характеристик нескольких базовых моделей. Если изменение конфигурации петель модифицированной ИОЛ может привести к потенциальному увеличению риска осложнений по сравнению с базовой моделью, то такую модификацию подвергают испытаниям не менее чем на 300 субъектах.
С.2.3.3 Изменения материала или калибра петель
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Изменение материала петель (или другого материала части базовой модели) или калибра петель может привести к созданию модификации ИОЛ, не соответствующей требованиям С.2.2.5 при ее сравнении с базовой моделью или с рядом характеристик нескольких базовых моделей.
С.2.3.4 Изменение материала петель
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Изменение материала петель ИОЛ на материал, не известный испытателю, но используемый для петель по данным офтальмологических исследований (статей, отчетов), должно быть подтверждено схожестью свойств используемых материалов.
С.2.3.5 Изменение материала тела
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1.
Изменение материала тела ИОЛ на материал, не известный испытателю, но используемый для тела ИОЛ по данным офтальмологических исследований (статей, отчетов), должно быть подтверждено схожестью свойств используемых материалов.
С.2.3.6 Изменение диаметра тела или оптической зоны
Признаки модифицированных ИОЛ типов P/SS/SN должны соответствовать указанным в С.2.1.1 при изменении диаметра тела или оптической части в интервале от 5,0 до 7,5 мм. Оценка модели ИОЛ с оптической частью диаметром менее 5 мм должна включать клинические испытания по оценке влияния бликов на остроту зрения пациентов.
С.2.3.7 Начальное добавление модели рубцом
Признаки модифицированных ИОЛ типов Р должны соответствовать указанным в С.2.1.1 при дополнении модели рубцом на задней поверхности тела ИОЛ.
Приложение D
(рекомендуемое)
Элементы, включаемые в протокол клинических испытаний ИОЛ,
и дополнительное клиническое руководство по разработке плана
клинических испытаний и анализу результатов
D.1 Элементы, включаемые в протокол клинических испытаний ИОЛ
D.1.1 Общие положения
Настоящее приложение устанавливает наиболее важные элементы, включаемые в протокол клинических испытаний, необходимые организатору для определения безопасности и эффективности ИОЛ.
D.1.2 Контрольная группа
Клинические результаты по исторической контрольной группе приведены в приложении А. Клиническое действие испытуемых ИОЛ сравнивают с данными, приведенными в приложении А или с данными одновременно прослеживаемой контрольной группы.
D.1.3 Число субъектов
D.1.3.1 При сравнении клинического действия ИОЛ с исторической контрольной группой клиническим испытаниям подвергают не менее 300 субъектов. При сравнении клинического действия испытуемых ИОЛ с клиническим действием ИОЛ одновременно испытуемой контрольной группой организатор должен подобрать такое число субъектов, которое будет достаточным для определения изменений остроты зрения и неблагоприятных исходов для сравнения с 300 субъектами исторической контрольной группы.
D.1.3.2 Для компенсации субъектов, которые могут быть не доведены до конца испытаний, рекомендуется использовать:
- при одногодичных испытаниях - 390 субъектов;
- при трехгодичных испытаниях - 500 субъектов.
Организатор не должен проводить клинические испытания на большем числе субъектов, чтобы число субъектов, подвергаемых риску имплантации моделей ИОЛ, которые еще не были признаны безопасными и эффективными, было минимальным.
D.1.3.3 Для обеспечения баланса числа субъектов у каждого испытателя организатор должен предусмотреть наличие не менее 20 субъектов, но не более 25% общего числа субъектов.
D.1.3.4 Чтобы достоверность данных была максимальной, число не доведенных до конца субъектов не должно превышать 30% при трехгодичных испытаниях и 10% - при одногодичных испытаниях.
D.1.4 Длительность клинических испытаний
D.1.4.1 Для оценки числа неблагоприятных исходов клинические испытания проводят в течение:
- одного года - для заднекамерных ИОЛ (форма отчета в соответствии с таблицей D.5 приложения D);
- трех лет - для переднекамерных ИОЛ (форма отчета в соответствии с таблицей D.7 приложения D).
Это применимо в случае модификации поверхности ИОЛ с нанесением покрытий или химической обработкой для увеличения биологической совместимости ИОЛ.
D.1.4.2 Для сведения риска, связанного с клиническими испытаниями новых моделей ИОЛ, к минимуму испытания проводят в два этапа:
1 - имплантация модели ИОЛ не более чем 100 субъектам. После того как по 50 субъектам будет получен отчет по форме отчета в соответствии с таблицей D.4 приложения D, результаты должны быть клинически оценены организатором и испытателем. В случае, если модель ИОЛ у данных 50 субъектов будет признана приемлемой, организатор может начать следующий этап испытаний.
2 - имплантация модели ИОЛ достаточному для окончания испытаний числу субъектов.
D.1.5 Отчетные периоды
D.1.5.1 Результаты клинических испытаний, проводимых в течение одного года, представляют в виде следующих отчетов:
- 0 - предоперационный/операционный отчет (форма отчета в соответствии с таблицами D.1 и/или D.3 приложения D);
- 1 - послеоперационный отчет через 1 или 2 дня после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D);
- 2 - послеоперационный отчет с 7-го по 14-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D);
- 3 - послеоперационный отчет с 30-го по 60-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D);
- 4 - послеоперационный отчет со 120-го по 180-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D);
- 5 - послеоперационный отчет с 330-го по 420-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D).
D.1.5.2 Результаты трехгодичных клинических испытаний представляют в виде отчетов по D.1.5.1, а также следующих:
- 6 - послеоперационный отчет с 630-го по 780-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D);
- 7 - послеоперационный отчет с 990-го по 1140-й день после операции (форма отчета в соответствии с таблицами D.2 и/или D.4 приложения D).
Для сбора информации о возможных неблагоприятных исходах необходимо проводить клиническую оценку данных по этим отчетным периодам.
D.1.5.3 Незапланированные посещения субъектов и процедуры для устранения неблагоприятных исходов, проводимые вне отчетных периодов, также должны быть занесены в отчет клинических испытаний.
D.1.5.4 За каждый отчетный период трехгодичных и одногодичных клинических испытаний должно быть получено не менее 300 отчетов.
D.2 Дополнительное клиническое руководство по разработке плана клинических испытаний и анализу результатов
D.2.1 Общие положения
Дополнительное клиническое руководство по разработке плана клинических испытаний и анализу результатов приведено в D.2.2-D.2.4.
D.2.2 Стандартизация клинических оценок
D.2.2.1 Организатор должен обеспечить соответствие критериев клинической оценки, полученных испытателями для неблагоприятных исходов и контроля параметров, относящихся к остроте зрения (например, конструкции таблицы оптотипов, уровня освещенности, отражательной способности таблицы, расстояния до таблицы, общей методологии), чтобы данные исследований могли быть скомбинированы.
D.2.2.2 Если организатор выявляет необходимость приведения оптотипов, использованных при проведении клинических испытаний, к стандартным оптотипам, то используют методику [3].
D.2.3 Руководство по анализу данных
D.2.3.1 Для выявления наличия или отсутствия проблем в конструкции ИОЛ, не установленных при общем анализе неблагоприятных исходов и остроты зрения (далее - ОЗ), организатор должен провести клиническую оценку данных по следующим показателям:
- ОЗ по возрасту;
- лучшие результаты ОЗ;
- ОЗ при неблагоприятных исходах;
- ОЗ по предоперационной окулярной патологии;
- анализ причин недостижения ОЗ 0,5 (6/12; 20/40) отн. ед. по каждому пациенту;
- соотношения нарастающих неблагоприятных исходов по возрасту;
- соотношения повторяющихся неблагоприятных исходов по возрасту;
- соотношения других неблагоприятных исходов;
- ОЗ у испытателей;
- неблагоприятные исходы по испытателям.
Такая клиническая оценка данных поможет организатору определить, зависит ли неблагоприятный исход от испытуемой ИОЛ или он вызван другими факторами.
D.2.3.2 Организатор должен помнить о том, что клиническое воздействие для исторических контрольных групп рассмотрено только для неблагоприятных исходов, приведенных в приложении А. Организатор должен использовать клинические оценки собственных исследований, данные из публикаций и/или данные из проведенных ранее клинических испытаний, если они существуют, в случае наличия других неблагоприятных исходов.
D.2.4 Клинические отчетные формы
Примеры отчетных форм приведены в таблицах D.1-D.5:
Таблица D.1 - Форма предоперационного отчета по оценке заднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения |
||||||||||||
|
день, месяц, год |
||||||||||||
|
Предоперационный отчет | |||||||||||||||||||||||||||||
|
день, месяц, год |
|||||||||||||||||||||||||||||
|
|
Оперируемый глаз |
Здоровый глаз |
Катаракта: Этиология: | ||||||||||||||||||||||||||
|
|
старческая другая |
? | |||||||||||||||||||||||||||
|
число пальцев |
? |
? |
указать вид |
||||||||||||||||||||||||||
|
движение руки |
? |
? |
Патология: |
Да |
Нет |
Не определяется | |||||||||||||||||||||||
|
реакция на свет |
? |
? |
псевдоэксфолиации |
? |
? |
||||||||||||||||||||||||
|
нет реакции на свет |
? |
? |
глаукома |
? |
? |
||||||||||||||||||||||||
|
Внутриглазное давление, мм рт.ст: |
ранее оперированная глаукома |
? |
? |
||||||||||||||||||||||||||
|
оперируемый глаз |
здоровый глаз |
||||||||||||||||||||||||||||
|
|
плохое сужение зрачка |
? |
? |
||||||||||||||||||||||||||
|
Используется омывающий раствор: Да ? Нет ? |
перенесенные уветиты |
? |
? |
||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||
|
|
указать наименование |
перенесенная отслойка сетчатки |
? |
? |
|||||||||||||||||||||||||
|
Внутриокулярные медикаменты: |
диабетическая ретинопатия |
? |
? |
||||||||||||||||||||||||||
|
Да |
Нет |
макулярная дегенерация |
? |
? |
|||||||||||||||||||||||||
|
антибиотик |
? |
? |
амблиопия |
? |
? |
? | |||||||||||||||||||||||
|
|
|
наименование антибиотика |
|
| |||||||||||||||||||||||||
|
|
наименование кортикостероида |
указать вид патологии |
|||||||||||||||||||||||||||
|
указать вид внутриокулярного медикамента |
Биометрия: | ||||||||||||||||||||||||||||
|
Разрез: |
К1, дптр |
длина оси, мм | |||||||||||||||||||||||||||
|
размер разреза, мм |
К2, дптр |
||||||||||||||||||||||||||||
|
расположение |
предполагаемая послеоперационная | ||||||||||||||||||||||||||||
|
роговичный, лимбальный, склеральный |
рефракция, дптр | ||||||||||||||||||||||||||||
|
Состояние роговицы: |
Расписка об ознакомлении получена: Да ? | ||||||||||||||||||||||||||||
|
Да |
Нет |
||||||||||||||||||||||||||||
|
нормальное |
? |
? |
день, месяц, год |
||||||||||||||||||||||||||
|
эндотелиальная дистрофия |
? |
? |
|||||||||||||||||||||||||||
|
другая |
|||||||||||||||||||||||||||||
|
указать вид патологии |
|||||||||||||||||||||||||||||
Таблица D.2 - Форма операционного отчета по оценке заднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения |
||||||||||||
|
день, месяц, год |
||||||||||||
|
Операционный отчет |
Дата операции | |||||||||||||||||
|
день, месяц, год |
день, месяц, год | |||||||||||||||||
|
Применен вискоэластик |
Да ? |
Нет ? |
Другие хирургические процедуры: | |||||||||||||||
|
Если применен, указать вид |
Да |
Нет | ||||||||||||||||
|
|
|
| ||||||||||||||||
|
Интраокулярные медикаменты: |
||||||||||||||||||
|
Да |
Нет |
иридотомия/иридоэктомия |
? |
? | ||||||||||||||
|
адреналин |
? |
? |
сфинктеротомия |
? |
? | |||||||||||||
|
ацетилхолин |
? |
? |
другая | |||||||||||||||
|
|
|
|
указать вид процедуры | |||||||||||||||
|
|
Проблемы при операции: | |||||||||||||||||
|
указать вид медикамента |
Да ? |
Нет ? | ||||||||||||||||
|
Вид экстракции хрусталика: |
кровотечение в переднем отделе |
? |
? | |||||||||||||||
|
плановая экстракапсулярная экстракция катаракты |
? |
повреждение радужки |
? |
? | ||||||||||||||
|
факоэмульсификация |
? |
остаточная непрозрачность задней капсулы |
? |
? | ||||||||||||||
|
другая |
разрыв задней капсулы |
? |
? | |||||||||||||||
|
указать вид |
|
|||||||||||||||||
|
Вид капсулотомии: |
||||||||||||||||||
|
непрерывный капсула рексис |
? |
указать вид проблемы |
||||||||||||||||
|
по типу "консервной банки" |
? |
|||||||||||||||||
|
другая |
? |
|||||||||||||||||
|
указать вид |
||||||||||||||||||
|
Расположение петель: |
Причина, по которой ИОЛ не была имплантирована: | |||||||||||||||||
|
в сумке |
? |
Место для наклейки этикетки имплантированной ИОЛ | ||||||||||||||||
|
частично в сумке |
? |
Время с момента начала до завершения операции | ||||||||||||||||
|
в сужении |
? |
мин | ||||||||||||||||
|
не определено |
? |
|||||||||||||||||
|
подпись испытателя |
день, месяц, год |
|||||||||||||||||
Таблица D.3 - Форма послеоперационного отчета по оценке заднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | ||||||||||||
|
фамилия, имя, отчество |
код | ||||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | ||||||||
|
код |
|||||||||||||
|
женский |
? |
негроид |
? | ||||||||||
|
монголоид |
? | ||||||||||||
|
другая |
? | ||||||||||||
|
смешанная |
? | ||||||||||||
|
Дата рождения |
|||||||||||||
|
день, месяц, год |
|||||||||||||
|
Послеоперационный отчет | ||||||||||||||||||||||||||||||
|
день, месяц, год |
||||||||||||||||||||||||||||||
|
Глаз: |
правый ? |
левый ? |
Другая патология или осложнение: | |||||||||||||||||||||||||||
|
Пациент не доступен для обследования в назначенное время, но продолжает участвовать в испытании ? |
Наличие |
Отсутствие | ||||||||||||||||||||||||||||
|
выпадение стекловидного тела в переднюю камеру |
? |
? | ||||||||||||||||||||||||||||
|
Пациент прекращает участвовать в испытании |
||||||||||||||||||||||||||||||
|
указать причину |
ущемление стекловидного тела в ране |
? |
? | |||||||||||||||||||||||||||
|
повышение внутриглазного давления, требующее лечения |
? |
? | ||||||||||||||||||||||||||||
|
блокирован зрачок |
? |
? | ||||||||||||||||||||||||||||
|
передние синехии |
? |
? | ||||||||||||||||||||||||||||
|
задние синехии |
? |
? | ||||||||||||||||||||||||||||
|
Рефракция: |
воспалительные отложения на ИОЛ |
? |
? | |||||||||||||||||||||||||||
|
сфера |
фибрин в зрачке |
? |
? | |||||||||||||||||||||||||||
|
цилиндр |
кортикальные остатки |
? |
? | |||||||||||||||||||||||||||
|
ось |
остатки ядра |
? |
? | |||||||||||||||||||||||||||
|
Кератометрия: |
децентрировка оптики ИОЛ |
? |
? | |||||||||||||||||||||||||||
|
К1, дптр |
мм |
|||||||||||||||||||||||||||||
|
указать размер |
||||||||||||||||||||||||||||||
|
К2, дптр |
||||||||||||||||||||||||||||||
|
отслойка сетчатки |
? |
? | ||||||||||||||||||||||||||||
|
диабетическая ретинопатия |
? |
? | ||||||||||||||||||||||||||||
|
цистоидный отек макулы |
? |
? | ||||||||||||||||||||||||||||
|
Оперируемый глаз |
Здоровый глаз |
в случае наличия - метод диагностики: |
||||||||||||||||||||||||||||
|
Лучшая скорригированная |
клинический |
|||||||||||||||||||||||||||||
|
|
флюоресцентная ангиография |
|||||||||||||||||||||||||||||
|
число пальцев |
? |
? |
макулярная дегенерация |
? |
? | |||||||||||||||||||||||||
|
движение руки |
? |
? |
атрофия радужки
|
? |
? | |||||||||||||||||||||||||
|
указать вид патологии |
||||||||||||||||||||||||||||||
|
нет реакции на свет |
? |
? |
||||||||||||||||||||||||||||
|
Внутриглазное давление, мм рт.ст. |
Затронута ли передняя камера: |
|||||||||||||||||||||||||||||
|
Да |
Нет | |||||||||||||||||||||||||||||
|
если не затронута: |
||||||||||||||||||||||||||||||
|
фиброз задней камеры |
? |
? | ||||||||||||||||||||||||||||
|
шары Эльшнига |
? |
? | ||||||||||||||||||||||||||||
|
если затронута: |
||||||||||||||||||||||||||||||
|
открывалась ли капсула с предыдущего визита |
? |
? | ||||||||||||||||||||||||||||
|
Используемые на момент визита медикаменты: |
Основная причина, по которой острота зрения менее 0,5: | |||||||||||||||||||||||||||||
|
Временно |
Систематически |
|||||||||||||||||||||||||||||
|
Да |
Нет |
Да |
Нет |
|||||||||||||||||||||||||||
|
кортикостероиды |
? |
? |
? |
? |
||||||||||||||||||||||||||
|
антибиотики |
? |
? |
? |
? |
||||||||||||||||||||||||||
|
нестероидные противовоспалительные средства |
? |
? |
? |
? |
||||||||||||||||||||||||||
|
глаукомные препараты |
? |
? |
? |
? |
||||||||||||||||||||||||||
|
другие |
||||||||||||||||||||||||||||||
|
указать наименования |
||||||||||||||||||||||||||||||
|
Раздражение: |
Были ли повторные хирургические вмешательства на оперированном глазу с прошлого визита: |
Да ? |
Нет ? | |||||||||||||||||||||||||||
|
нет |
? |
|||||||||||||||||||||||||||||
|
легкое |
? |
Если были, указать какие | ||||||||||||||||||||||||||||
|
умеренное |
? |
|||||||||||||||||||||||||||||
|
тяжелое |
? |
|||||||||||||||||||||||||||||
|
Другая патология или осложнение: |
Наличие |
Отсутствие |
Испытывал ли пациент неблагоприятное воздействие: | |||||||||||||||||||||||||||
|
фильтрация из раны |
? |
? |
Да |
Нет | ||||||||||||||||||||||||||
|
плоская передняя камера |
? |
? |
? |
? | ||||||||||||||||||||||||||
|
гифема, мм |
? |
? |
||||||||||||||||||||||||||||
|
указать размер |
Если испытывал, заполнить форму отчета о неблагоприятном воздействии. | |||||||||||||||||||||||||||||
|
Если воздействие серьезное, заполнить форму отчета о неблагоприятном воздействии и связаться с организатором в течение одного рабочего дня | ||||||||||||||||||||||||||||||
Таблица D.4 - Форма предоперационного отчета по оценке переднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения | ||||||||||||
|
день, месяц, год |
||||||||||||
|
Предоперационный отчет | ||||||||||||||||||||||||
|
день, месяц, год |
||||||||||||||||||||||||
|
|
Оперируемый глаз |
Здоровый глаз |
Катаракта: этиология: | |||||||||||||||||||||
|
|
старческая другая |
? | ||||||||||||||||||||||
|
число пальцев |
? |
? |
указать вид |
|||||||||||||||||||||
|
движение руки |
? |
? |
Патология: |
Да |
Нет |
Не определяется | ||||||||||||||||||
|
реакция на свет |
? |
? |
псевдоэксфолиации |
? |
? |
|||||||||||||||||||
|
нет реакции на свет |
? |
? |
глаукома |
? |
? |
|||||||||||||||||||
|
Используется омывающий раствор: Да ? Нет ? |
ранее оперированная глаукома |
? |
? |
|||||||||||||||||||||
|
Если используется, указать наименование |
плохое сужение зрачка |
? |
? |
|||||||||||||||||||||
|
Внутриокулярные медикаменты: |
перенесенные уветиты |
? |
? |
|||||||||||||||||||||
|
|
Да |
Нет |
перенесенная отслойка сетчатки |
? |
? |
|||||||||||||||||||
|
наименование антибиотика |
|
|
|
|||||||||||||||||||||
|
кортикостероид |
? |
? |
||||||||||||||||||||||
|
наименование кортикостероида |
амблиопия |
? |
? |
? | ||||||||||||||||||||
|
другой |
другая |
|||||||||||||||||||||||
|
указать вид внутриокулярного медикамента |
указать вид патологии |
|||||||||||||||||||||||
|
Состояние роговицы: |
Расписка об ознакомлении получена: Да ? | |||||||||||||||||||||||
|
Да |
Нет |
|||||||||||||||||||||||
|
нормальное |
? |
? |
день, месяц, год |
|||||||||||||||||||||
|
эндотелиальная дистрофия |
? |
? |
||||||||||||||||||||||
|
другая |
||||||||||||||||||||||||
|
указать вид патологии |
||||||||||||||||||||||||
Таблица D.5 - Форма операционного отчета по оценке переднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения | ||||||||||||
|
день, месяц, год |
||||||||||||
|
Операционный отчет |
Дата операции | ||||||||||||||||||||||
|
день, месяц, год |
день, месяц, год | ||||||||||||||||||||||
|
Применен вискоэластик |
Да ? |
Нет ? |
Другие хирургические процедуры: | ||||||||||||||||||||
|
Если применен, указать вид |
Да |
Нет | |||||||||||||||||||||
|
Интраокулярные медикаменты: |
периферийная иридотомия/иридоэктомия |
? |
? | ||||||||||||||||||||
|
Да |
Нет |
сфинктеротомия |
? |
? | |||||||||||||||||||
|
адреналин |
? |
? |
другая |
? |
? | ||||||||||||||||||
|
указать вид процедуры |
|||||||||||||||||||||||
|
ацетилхолин |
? |
? |
|||||||||||||||||||||
|
карбахол |
? |
? |
Проблемы при операции: | ||||||||||||||||||||
|
другой |
Да |
Нет | |||||||||||||||||||||
|
указать вид медикамента |
|||||||||||||||||||||||
|
|
кровотечение в переднем отделе |
? |
? | ||||||||||||||||||||
|
повреждение радужки |
? |
? | |||||||||||||||||||||
|
остаточная непрозрачность задней капсулы |
? |
? | |||||||||||||||||||||
|
разрыв задней капсулы |
? |
? | |||||||||||||||||||||
|
передняя витрэктомия | |||||||||||||||||||||||
|
указать вид проблемы |
|||||||||||||||||||||||
|
Вид экстракции хрусталика: |
Причина, по которой ИОЛ не была имплантирована: | ||||||||||||||||||||||
|
плановая экстракапсулярная экстракция катаракты |
? |
||||||||||||||||||||||
|
факоэмульсификация |
? |
Место для наклейки этикетки имплантированной ИОЛ | |||||||||||||||||||||
|
другая |
|||||||||||||||||||||||
|
указать вид |
|||||||||||||||||||||||
|
Вид капсулотомии: |
Время с момента начала до завершения операции | ||||||||||||||||||||||
|
непрерывный капсула рексис |
? |
мин | |||||||||||||||||||||
|
по типу "консервной банки" |
? |
||||||||||||||||||||||
|
другая |
? |
||||||||||||||||||||||
|
указать вид |
|||||||||||||||||||||||
|
Расположение петель: |
|||||||||||||||||||||||
|
в сумке |
? |
подпись испытателя |
день, месяц, год |
||||||||||||||||||||
|
частично в сумке |
? |
||||||||||||||||||||||
|
в сужении |
? |
||||||||||||||||||||||
|
не определено |
? |
||||||||||||||||||||||
Таблица D.6 - Форма послеоперационного отчета по оценке переднекамерных ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения | ||||||||||||
|
день, месяц, год |
||||||||||||
|
Послеоперационный отчет | |||||||||||||||||||||||||||||
|
день, месяц, год |
|||||||||||||||||||||||||||||
|
Глаз: |
правый ? |
левый ? |
Другая патология или осложнение: | ||||||||||||||||||||||||||
|
Пациент не доступен для обследования в назначенное время, но продолжает участвовать в испытании |
? |
Наличие |
Отсутствие | ||||||||||||||||||||||||||
|
выпадение стекловидного тела в переднюю камеру |
? |
? | |||||||||||||||||||||||||||
|
Пациент прекращает участвовать в испытании |
|||||||||||||||||||||||||||||
|
указать причину |
ущемление стекловидного тела в ране |
? |
? | ||||||||||||||||||||||||||
|
Рефракция: |
повышение внутриглазного давления, требующее лечения |
? |
? | ||||||||||||||||||||||||||
|
блокирован зрачок |
? |
? | |||||||||||||||||||||||||||
|
цилиндр |
|||||||||||||||||||||||||||||
|
передние синехии |
? |
? | |||||||||||||||||||||||||||
|
ось |
|||||||||||||||||||||||||||||
|
задние синехии |
? |
? | |||||||||||||||||||||||||||
|
Кератометрия: |
воспалительные отложения на ИОЛ |
? |
? | ||||||||||||||||||||||||||
|
К1, дптр |
фибрин в зрачке |
? |
? | ||||||||||||||||||||||||||
|
К2, дптр |
кортикальные остатки |
? |
? | ||||||||||||||||||||||||||
|
Оперируемый глаз |
Здоровый глаз |
остатки ядра |
? |
? | |||||||||||||||||||||||||
|
Лучшая скорригированная |
децентрировка оптики ИОЛ |
? |
? | ||||||||||||||||||||||||||
|
мм |
|||||||||||||||||||||||||||||
|
Отметить нужное: |
указать размер |
||||||||||||||||||||||||||||
|
число пальцев |
? |
? |
отслойка сетчатки |
? |
? | ||||||||||||||||||||||||
|
движение руки |
? |
? |
диабетическая ретинопатия |
? |
? | ||||||||||||||||||||||||
|
реакция на свет |
? |
? |
цистоидный отек макулы |
? |
? | ||||||||||||||||||||||||
|
нет реакции на свет |
? |
? |
в случае наличия, метод диагностики: |
||||||||||||||||||||||||||
|
клинический |
|||||||||||||||||||||||||||||
|
флюоресцентная ангиография |
|||||||||||||||||||||||||||||
|
макулярная дегенерация |
? |
? | |||||||||||||||||||||||||||
|
атрофия радужки |
? |
? | |||||||||||||||||||||||||||
|
другая |
? |
? | |||||||||||||||||||||||||||
|
Внутриглазное давление, мм рт.ст. |
Затронута ли передняя камера: |
||||||||||||||||||||||||||||
|
Да |
Нет | ||||||||||||||||||||||||||||
|
Если не затронута: |
|||||||||||||||||||||||||||||
|
фиброз задней камеры |
? |
? | |||||||||||||||||||||||||||
|
шары Эльшнига |
? |
? | |||||||||||||||||||||||||||
|
Если затронута: |
|||||||||||||||||||||||||||||
|
открывалась ли капсула с предыдущего визита |
? |
? | |||||||||||||||||||||||||||
|
Используемые на момент визита медикаменты: |
Основная причина, по которой острота зрения менее 0,5: | ||||||||||||||||||||||||||||
|
Временно |
Систематически |
||||||||||||||||||||||||||||
|
Да |
Нет |
Да |
Нет |
||||||||||||||||||||||||||
|
кортикостероиды |
? |
? |
? |
? |
|||||||||||||||||||||||||
|
антибиотики |
? |
? |
? |
? |
|||||||||||||||||||||||||
|
нестероидные противовоспалительные средства |
? |
? |
? |
? |
|||||||||||||||||||||||||
|
глаукомные препараты |
? |
? |
? |
? |
|||||||||||||||||||||||||
|
другие |
|||||||||||||||||||||||||||||
|
указать наименования |
|||||||||||||||||||||||||||||
|
Раздражение: |
Были ли повторные хирургические вмешательства на оперированном глазу с прошлого визита |
Да |
Нет | ||||||||||||||||||||||||||
|
нет |
? |
? |
? | ||||||||||||||||||||||||||
|
легкое |
? |
Если были, указать какие | |||||||||||||||||||||||||||
|
умеренное |
? |
||||||||||||||||||||||||||||
|
тяжелое |
? |
||||||||||||||||||||||||||||
|
Другая патология или осложнение: |
Испытывал ли пациент неблагоприятное воздействие: | ||||||||||||||||||||||||||||
|
Наличие |
Отсутствие |
Да |
Нет | ||||||||||||||||||||||||||
|
фильтрация из раны |
? |
? |
? |
? | |||||||||||||||||||||||||
|
плоская передняя камера |
? |
? |
Если испытывал, заполнить форму отчета о неблагоприятном воздействии. | ||||||||||||||||||||||||||
|
гифема, мм |
? |
? |
|||||||||||||||||||||||||||
|
указать размер |
Если воздействие серьезное, заполнить форму отчета о неблагоприятном воздействии и связаться с организатором в течение одного рабочего дня | ||||||||||||||||||||||||||||
Таблица D.7 - Форма отчета о неблагоприятном воздействии ИОЛ
|
Испытатель |
Клиническое испытание | |||||||||||
|
фамилия, имя, отчество |
код | |||||||||||
|
Пациент |
Инициалы пациента |
Пол: мужской |
? |
Раса: европеоид |
? | |||||||
|
код |
||||||||||||
|
женский |
? |
негроид |
? | |||||||||
|
монголоид |
? | |||||||||||
|
другая |
? | |||||||||||
|
смешанная |
? | |||||||||||
|
Дата рождения | ||||||||||||
|
день, месяц, год |
||||||||||||
|
Испытатель обязан сообщать о любых неблагоприятных воздействиях, связанных либо не связанных с испытуемой ИОЛ. О любых серьезных и/или неожиданных реакциях необходимо в течение одного рабочего дня доложить организатору по телефону и/или факсу. В случае серьезного неблагоприятного воздействия форма отчета о неблагоприятном исходе должна быть заполнена, подписана и передана организатору в течение пяти рабочих дней. | ||||||||||||||||||||||||
|
1 Оперированный глаз: правый (OD) |
? |
11 Лечение неблагоприятного воздействия: | ||||||||||||||||||||||
|
левый (OS) |
? |
12 Исход: | ||||||||||||||||||||||
|
2 Дата имплантации |
полное восстановление |
? |
||||||||||||||||||||||
|
день, месяц, год |
|
|
||||||||||||||||||||||
|
3 Код модели линзы |
||||||||||||||||||||||||
|
воздействие продолжается на момент отчета |
? |
|||||||||||||||||||||||
|
4 Серийный номер линзы |
||||||||||||||||||||||||
|
смерть |
? |
|||||||||||||||||||||||
|
5 Оптическая сила линзы, дптр |
||||||||||||||||||||||||
|
Неблагоприятное воздействие: |
||||||||||||||||||||||||
|
6 Дата обнаружения |
Прогноз в случае продолжения воздействия: | |||||||||||||||||||||||
|
день, месяц, год |
||||||||||||||||||||||||
|
7 Продолжительность (ч, дни, др.) |
||||||||||||||||||||||||
|
8 Тяжесть неблагоприятного воздействия: |
Резюме (печатными буквами): | |||||||||||||||||||||||
|
легкое |
? |
|||||||||||||||||||||||
|
умеренное |
? |
|||||||||||||||||||||||
|
тяжелое |
? |
|||||||||||||||||||||||
|
9 Описание течения неблагоприятного воздействия (печатными буквами): |
13 Считает ли заполняющий отчет, что эффект связан с линзой: | |||||||||||||||||||||||
|
Да |
Нет |
|||||||||||||||||||||||
|
? |
? |
|||||||||||||||||||||||
|
Резюме (печатными буквами): | ||||||||||||||||||||||||
|
Да |
Нет |
|||||||||||||||||||||||
|
? |
? |
|||||||||||||||||||||||
|
Если является, заполнить форму отчета и связаться с организатором в течение одного рабочего дня | ||||||||||||||||||||||||
|
10 Диагноз неблагоприятного воздействия (печатными буквами): |
||||||||||||||||||||||||
|
подпись испытателя |
день, месяц, год |
|||||||||||||||||||||||
|
Телефон | ||||||||||||||||||||||||
|
код |
номер |
дополнительный |
||||||||||||||||||||||
|
Приложение Е Окончательный отчет о клинических испытаниях ИОЛ Е.1 Общие положения |
Приложение F
(справочное)
Сопоставление структуры настоящего стандарта со структурой
примененного в нем международного стандарта
F.1 Общие положения
Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта представлено в таблице F.1. Указанное в таблице изменение структуры межгосударственного стандарта относительно структуры примененного в нем международного стандарта обусловлено приведением его в соответствие с требованиями ГОСТ 1.5-2004*.
________________
* На территории Российской Федерации действует ГОСТ Р 1.5-2004, здесь и далее по тексту. - Примечание .
Таблица F.1 - Сопоставление структуры настоящего стандарта со структурой примененного международного стандарта
|
Структура международного стандарта ИСО 11979-7:2001 |
Структура настоящего стандарта |
|
1 Обзор |
1 Область применения |
|
2 Нормативные ссылки |
2 Нормативные ссылки |
|
3 Термины и определения |
3 Термины и определения |
|
4 Этические соглашения |
4 Этические соглашения |
|
5 Требования |
5 Требования |
|
6 Методология |
6 Методология |
|
7 Представление результатов |
7 Требования к представлению результатов клинических испытаний |
|
Приложение А Избранные определения |
- |
|
Приложение В Примеры модификаций базовой модели ИОЛ |
Приложение С Руководство по определению модификации базовой модели ИОЛ |
|
Приложение С Элементы протоколирования клинических испытаний ИОЛ |
Приложение D Элементы, включаемые в протокол клинических испытаний ИОЛ, и дополнительное клиническое руководство по разработке плана клинических испытаний и анализу результатов |
|
Приложение D Послеоперационные неблагоприятные исходы и нормы остроты зрения в сравнении с исторической контрольной группой |
Приложение А Примеры серьезных неблагоприятных исходов для переднекамерных и заднекамерных ИОЛ и показателей остроты зрения |
|
- |
Приложение В Требования к расписке об информированности |
|
- |
Приложение Е Окончательный отчет о клинических испытаниях ИОЛ |
|
Библиография |
Приложение G Библиография |
|
- |
Приложение F Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта |
|
________________ | |
Приложение G
(справочное)
Библиография
|
[1] |
ИСО 14155-1:2003 |
Клинические испытания медицинских изделий. Часть 1. Общие требования |
|
(ISO 14155-1:2003) |
(Clinical investigation of medical device for human subjects - Part 1: General requirements) | |
|
[2] |
ИСО 14155-2:2003 |
Клинические испытания медицинских изделий. Часть 2. План клинических испытаний |
|
(ISO 14155-2:2003) |
(Clinical investigation of medical device for human subjects - Part 2: Clinical investigation plans) | |
|
[3] |
ИСО 8597:1994 |
Оптика и оптические приборы. Проверка остроты зрения. Метод корреляции оптотипов |
|
(ISO 8597:1994) |
(Optics and optical instruments. Visual acuity testing. Correlation of optotypes) |
Текст документа сверен по:
официальное издание
М.: Стандартинформ, 2006
Личный кабинет:
доступно после авторизации Кубанский блогер Каграманов рассказал в «Шоу Воли» о хейте в...
Кубанский блогер Каграманов рассказал в «Шоу Воли» о хейте в...  Создайте свой интернет-магазин на новой платформе ReadyScript
Создайте свой интернет-магазин на новой платформе ReadyScript  Хостинг, домены, VPS/VDS, размещение серверов
Хостинг, домены, VPS/VDS, размещение серверов
© 2007-2025 ООО «РуФокс»
о проекте
вакансии
хостинг
создание сайтов
реклама на сайте
наши партнеры
сообщить об ошибке