
- USD ЦБ 03.12 30.8099 -0.0387
- EUR ЦБ 03.12 41.4824 -0.0244
|
Краснодар:
|
погода |
декабря
18
среда,
Курсы
Индексы
- DJIA 03.12 12019.4 -0.01
- NASD 03.12 2626.93 0.03
- RTS 03.12 1545.57 -0.07
ГОСТ Р ИСО 14971-2006
Группа Р20
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИЗДЕЛИЯ МЕДИЦИНСКИЕ
Применение менеджмента риска к медицинским изделиям
Medical devices. Application of risk management to medical devices
ОКС 11.040.01
ОКП 94 4000
Дата введения 2007-01-01
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ "О техническом регулировании", а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 "Стандартизация в Российской Федерации. Основные положения"
Сведения о стандарте
1 ПОДГОТОВЛЕН Закрытым акционерным обществом "ВНИИМП-ВИТА" на основе аутентичного перевода стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 436 "Управление качеством и соответствующие аспекты для медицинских изделий"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 26 апреля 2006 г. N 78-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14971:2000 "Изделия медицинские. Применение менеджмента риска к медицинским изделиям" (ISO 14971:2000 "Medical devices - Application of risk management to medical devices") и изменению N 1 (ISО 14971:2000/Amd. 1:2003), которое в тексте стандарта выделено двойной вертикальной линией
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации, сведения о которых приведены в дополнительном приложении J
5 ВЗАМЕН ГОСТ Р ИСО 14971-1-99
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячно издаваемых информационных указателях "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
Введение
Международный стандарт ИСО 14971 разработан Техническим комитетом ИСО/ТК 210 "Менеджмент качества и соответствующие общие аспекты медицинских изделий" и подкомитетом МЭК/ПК 62А "Общие аспекты электрических изделий, применяемых в медицинской практике". В области менеджмента риска применительно к медицинским изделиям Технический комитет ИСО/ТК 210 и МЭК/ПК 62А образовали совместную рабочую группу СРГ 1 "Применение менеджмента риска к медицинским изделиям".
Требования, предъявляемые к компоненту анализа риска как составляющей процесса менеджмента риска, были впервые разработаны в стандарте ИСО 14971-1:1998, причем было предусмотрено, что требования к оцениванию риска, управлению риском и к оцениванию постпроизводственной информации могут быть рассмотрены в дополнительной части (частях). Однако все эти требования включены в настоящий стандарт.
Настоящее издание ИСО 14971 отменяет и заменяет ИСО 14971-1:1998.
Подкомитет МЭК/ПК 62А принял решение, что настоящая публикация действует до 2004 года. По истечении этого срока подкомитет МЭК/ПК 62А после консультации с ИСО/ТК 210 решит, будет ли настоящая публикация:
- подтверждена;
- отозвана;
- заменена пересмотренным изданием;
- дополнена изменениями.
В ожидании пересмотра настоящего стандарта СРГ 1 разработала изменение к настоящей публикации, в котором документировано обоснование требований, содержащихся в стандарте ИСО 14971:2000, и которое включено в настоящий стандарт в качестве приложения Н.
Приложения А-Н настоящего стандарта являются справочными.
Настоящий стандарт рекомендуется рассматривать как основу для осуществляемого изготовителем результативного менеджмента всех рисков, связанных с применением медицинских изделий. Содержащиеся в настоящем стандарте требования составляют систему, в рамках которой практический опыт, интуицию и расчет используют для менеджмента вышеуказанных рисков.
Виды деятельности, в которых участвуют отдельные лица, организации или органы государственного управления, могут стать для этих или других участвующих сторон источником опасностей, способных вызвать потерю или причинить ущерб. Менеджмент рисков представляет собой сложную для обсуждения тему, так как каждая из участвующих сторон может по-разному оценивать вероятность причинения вреда и возможность нанесения ущерба в случае опасности.
Концепция риска включает в себя два компонента:
a) вероятность причинения вреда, то есть возможную частоту его нанесения;
b) последствия причиненного вреда, то есть его тяжесть.
Эти компоненты определяют допустимость риска для участвующей стороны.
Рассмотренная концепция особенно важна применительно к медицинским изделиям из-за большого разнообразия участвующих сторон, включая врачей-практиков, медицинские организации, органы государственного управления, промышленность, пациентов и общественность.
Все участвующие стороны должны понимать, что применение медицинских изделий связано с определенными рисками. Факторы, влияющие на восприятие рисков каждой участвующей стороной, включают в себя социально-экономический и образовательный статус заинтересованной части общества, а также объективно и субъективно оцениваемое состояние здоровья пациента. Характер восприятия рисков зависит, например, от того, рассматривают ли риски как:
- непроизвольные, неизбежные, обусловленные человеческим фактором;
- следствие небрежности;
- вызванные непонятной причиной или направленные на уязвимую группу лиц в обществе.
Решение о проведении клинической процедуры с применением медицинского изделия требует, чтобы остаточные риски были соотнесены с ожидаемой пользой от применения этой процедуры. При вынесении таких решений рекомендуется учитывать предусмотренное применение/предусмотренное назначение, эксплуатационные свойства и риски, связанные с конкретным медицинским изделием, а также риски и пользу, связанные с клинической процедурой или обстоятельствами ее применения. Некоторые из этих решений могут быть приняты только квалифицированным врачом-практиком, знающим состояние здоровья конкретного пациента или субъективное мнение пациента по этому вопросу.
В качестве одной из участвующих сторон изготовителю следует дать заключение о безопасности медицинского изделия, включающее допустимость рисков, с учетом общепризнанного уровня техники для определения пригодности медицинского изделия в целях размещения его на рынке в соответствии с предусмотренным применением/предусмотренным назначением. Настоящий стандарт устанавливает процедуру, с помощью которой изготовитель медицинского изделия может идентифицировать опасности, связанные с медицинским изделием и его принадлежностями, определять и оценивать риски, связанные с этими опасностями, управлять этими рисками и осуществлять мониторинг результативности такого управления.
1 Область применения
Настоящий стандарт устанавливает процедуру определения опасностей, связанных с медицинскими изделиями и их принадлежностями, включая изделия для in vitro диагностики, определения и оценивания рисков, управления рисками и мониторинга результативности такого управления.
Требования настоящего стандарта применимы ко всем этапам жизненного цикла медицинских изделий.
Настоящий стандарт не предназначен для вынесения клинических суждений, связанных с применением медицинских изделий.
Настоящий стандарт не устанавливает уровни допустимого риска.
Настоящий стандарт не требует наличия у изготовителя официально зарегистрированной системы качества. Однако менеджмент риска может быть составной частью системы качества (таблица G.1).
2 Термины и определения
В настоящем стандарте применяют следующие термины с соответствующими определениями:
2.1 эксплуатационный документ (accompanying document): Документ, прилагаемый к медицинскому изделию или принадлежности, содержащий всю важную информацию для пользователя, оператора, лица, производящего сборку или установку изделия на месте эксплуатации, в частности относящуюся к безопасности.
Примечание - На основании [1], подпункт 2.1.4.
2.2 вред (harm): Физическая травма или ущерб здоровью людей или имуществу, или окружающей среде ([2], пункт 3.1).
2.3 опасность (hazard): Потенциальный источник вреда ([2], пункт 3.5).
2.4 опасная ситуация (hazardous situation): Обстоятельства, при которых люди, имущество или окружающая среда подвержены одной или нескольким опасностям ([2], пункт 3.6).
2.5 предусмотренное применение/предусмотренное назначение (intended use/intended purpose): Применение изделия, процесса или услуги в соответствии с техническими условиями, инструкциями и информацией, предоставленными изготовителем.
2.6 изготовитель (manufacturer): Физическое или юридическое лицо, несущее ответственность за проектирование, изготовление, упаковывание и/или маркирование медицинского изделия, сборку системы или адаптирование медицинского изделия перед его введением в обращение и/или вводом в эксплуатацию, независимо от того, выполняет ли эти операции само вышеупомянутое лицо или третья сторона от его имени.
2.7 медицинское изделие (medical device): Прибор, аппарат, инструмент, устройство, комплект, комплекс, система с программными средствами, оборудование, приспособление, перевязочное и шовное средство, стоматологический материал, набор реагентов, контрольный материал и стандартный образец, изделие из полимерных, резиновых и иных материалов, применяемые в медицинских целях по отдельности или в сочетании друг с другом и предназначенные изготовителем для:
- профилактики, диагностики, лечения заболеваний, проведения медицинских процедур, исследований медицинского характера, замены или модификации частей тела человека, восстановления или компенсации нарушенных или утраченных физиологических функций, контроля над зачатием;
- воздействия на организм человека, при котором их функциональное назначение не реализуется, но может поддерживаться химическим, фармакологическим, иммунологическим или метаболическим взаимодействием с организмом человека ([3], пункт 3.1).
2.8 объективное свидетельство (objective evidence): Информация, правильность которой может быть доказана на основании фактов, полученных путем наблюдения, измерения, испытания или другими способами ([4], пункт 2.19).
2.9 процедура (procedure): Установленный способ осуществления какого-либо вида деятельности или процесса ([4], подпункт 3.4.5).
2.10 процесс (process): Совокупность взаимосвязанных видов деятельности и ресурсов, преобразующая входы в выходы ([4], подпункт 3.4.1).
2.11 запись (record): Документ, предоставляющий объективное свидетельство о выполненных видах деятельности или полученных результатах ([4], подпункт 3.7.6).
2.12 остаточный риск (residual risk): Риск, остающийся после предпринятых защитных мер ([2], пункт 3.9).
2.13 риск (risk): Сочетание вероятности причинения вреда и тяжести этого вреда ([2], пункт 3.2).
2.14 анализ риска (risk analysis): Систематическое использование имеющейся информации для выявления опасностей и определения риска ([2], пункт 3.10).
2.15 оценка риска (risk assessment): Полный процесс анализа и оценивания риска ([2], пункт 3.12).
2.16 управление риском (risk control): Процесс принятия решений и осуществления защитных мер по уменьшению рисков до установленных уровней или поддержания рисков на установленных уровнях.
2.17 оценивание риска (risk evaluation): Принятие решения, базирующегося на существующих общественных ценностях, о допустимости риска в конкретной ситуации на основании анализа риска.
Примечание - На основании [2], пункты 3.11 и 3.7.
2.18 менеджмент риска (risk management): Систематическое применение политики, процедур и практических методов менеджмента для решения задач анализа, оценивания и управления риском.
2.19 файл менеджмента риска (risk management file): Совокупность записей и других документов, не обязательно взаимосвязанных, создаваемых в процессе менеджмента риска.
2.20 безопасность (safety): Отсутствие недопустимого риска ([2], пункт 3.1).
2.21 тяжесть (severity): Степень возможных последствий опасности.
2.22 верификация (verification): Подтверждение на основе анализа и представления объективных свидетельств того, что установленные требования выполнены.
Примечание - При проектировании и разработке верификация означает процесс анализа результатов предпринятой деятельности с целью определения соответствия установленным к этой деятельности требованиям ([4], подпункт 3.8.4).
3.1 Национальные или региональные регулирующие требования
Ввиду большого разнообразия медицинских изделий, на которые распространяется действие настоящего стандарта, и различия национальных или региональных регулирующих требований, предъявляемых к медицинским изделиям, требования 3.3 и 3.4 применяют при необходимости.
3.2 Процесс менеджмента риска
Изготовитель должен устанавливать и поддерживать в рабочем состоянии процесс идентификации опасностей, связанных с медицинским изделием, определения и оценивания сопутствующих рисков, управления этими рисками и мониторинга результативности такого управления. Этот процесс должен быть документирован и включать в себя следующие элементы:
- анализ рисков;
- оценивание рисков;
- управление рисками;
- постпроизводственную информацию.
При наличии документированного процесса проектирования/разработки изделия в него должны входить соответствующие части процесса менеджмента риска.
Примечания
1 Документированный процесс проектирования/разработки может быть использован для обеспечения систематического подхода к рассмотрению проблем безопасности, позволяющего, в частности, выявлять на ранних стадиях опасности, связанные со сложными системами и окружающей средой.
2 Схема процесса менеджмента риска представлена на рисунке 1.
3 См. Библиографию.

Рисунок 1 - Схема процесса менеджмента риска
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
3.3 Ответственность высшего руководства
Высшее руководство организации-изготовителя должно:
a) разрабатывать политику определения допустимого риска с учетом соответствующих международных стандартов, а также национальных и региональных регламентов;
b) обеспечивать снабжение необходимыми ресурсами;
c) обеспечивать назначение обученного персонала (см. 3.4) для осуществления менеджмента, выполнения работ и получения оценки предпринимаемых действий;
d) проводить анализ результатов предпринимаемых действий по менеджменту риска через запланированные интервалы времени с цепью обеспечения постоянной пригодности и результативности процесса менеджмента риска.
Все изложенное в настоящем пункте должно быть задокументировано в файле менеджмента риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
3.4 Квалификация персонала
Изготовитель должен обеспечивать включение в состав персонала, выполняющего задачи менеджмента риска, лиц, имеющих знания и опыт, соответствующие выполнению поставленных задач. При необходимости эта квалификация должна включать в себя знание медицинских изделий и опыт их эксплуатации, а также владение методами менеджмента риска. Записи о соответствующей квалификации персонала необходимо поддерживать в рабочем состоянии.
Соответствие настоящему пункту проверяют контролем вышеуказанных записей.
3.5 План менеджмента риска
Изготовитель должен составлять план менеджмента риска в соответствии с процессом менеджмента риска для конкретных медицинского изделия или рассматриваемой принадлежности. План менеджмента риска должен быть частью файла менеджмента риска.
Этот план должен включать в себя:
a) область применения плана, идентификацию и описание медицинского изделия, а также стадии жизненного цикла изделия, к которым этот план применим;
b) план верификации;
c) распределение ответственности;
d) требования к анализу предпринимаемых действий по менеджменту риска;
e) критерии допустимости риска.
Примечание - Критерии допустимости риска имеют большое значение для определения конечной результативности процесса менеджмента риска. Руководство по определению таких критериев приведено в приложении Е.
При изменении плана в течение жизненного цикла медицинского изделия в файл менеджмента риска необходимо внести запись об изменении.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
3.6 Файл менеджмента риска
Для конкретных медицинского изделия или рассматриваемой принадлежности результаты всей деятельности по менеджменту риска записывают и поддерживают в рабочем состоянии в файле менеджмента риска.
Примечания
1 Записи и другие документы, составляющие файл менеджмента риска, могут быть частью других документов и файлов, необходимых, например, в системе менеджмента качества изготовителя.
2 Файл менеджмента риска не обязательно должен физически включать в себя все документы, относящиеся к настоящему стандарту. Однако он должен содержать, по меньшей мере, ссылки или указатели на все требуемые документы. Изготовителю рекомендуется своевременно собрать ссылочную информацию в файле менеджмента риска.
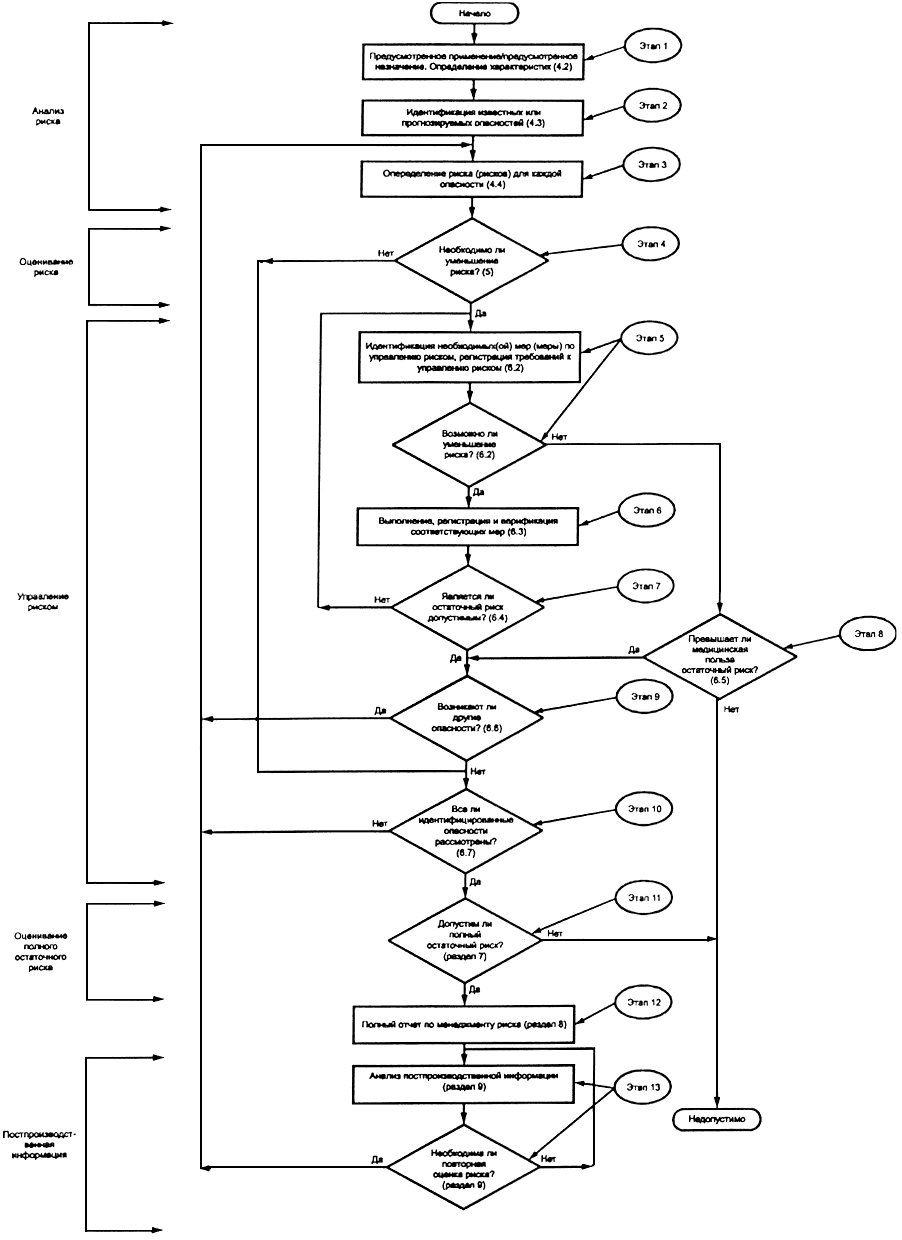
4.1 Процедура анализа риска
Анализ риска следует проводить в соответствии с 4.2-4.4, а порядок проведения и результаты анализа риска должны быть записаны в файле менеджмента риска.
Примечание - Если доступны результаты анализа риска для аналогичного медицинского изделия, то на них можно ссылаться при условии возможности демонстрации того, что рассматриваемые процессы аналогичны или внесенные изменения не приведут к существенным различиям в результатах. Основанием для этого должно быть систематическое оценивание изменений и возможностей их воздействия на различные существующие опасности.
В дополнение к записям в соответствии с 4.2-4.4, документы о порядке проведения и результатов анализа риска должны включать в себя, по меньшей мере:
a) описание и идентификацию анализируемого медицинского изделия или принадлежности;
b) идентификацию лица (лиц) и организации, выполняющих анализ риска;
c) дату проведения анализа риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
4.2 Предусмотренное применение/предусмотренное назначение и определение характеристик, относящихся к безопасности медицинского изделия (этап 1)
Для конкретных медицинского изделия или рассматриваемой принадлежности изготовитель должен составить описание предусмотренного применения/предусмотренного назначения и любого обоснованно прогнозируемого неправильного применения. Изготовитель должен также составить перечень всех качественных и количественных характеристик, которые могут повлиять на безопасность медицинского изделия, и, если возможно, указать их предельно допустимые значения (см. примечание 1). Эти записи необходимо поддерживать в рабочем состоянии в файле менеджмента риска.
Примечания
1 Приложение А настоящего стандарта содержит вопросы, которые могут служить полезным руководством при составлении такого перечня.
2 Приложение В настоящего стандарта содержит дополнительные руководящие указания по методам анализа риска в отношении медицинских изделий для in vitro диагностики.
3 Приложение С настоящего стандарта содержит дополнительные руководящие указания по методам анализа риска в отношении токсикологических опасностей.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
4.3 Идентификация известных или прогнозируемых опасностей (этап 2)
Изготовитель должен составить перечень известных или прогнозируемых опасностей, связанных с медицинским изделием, как для условий нормальной эксплуатации, так и для аварийных ситуаций. Известные опасности должны быть идентифицированы. Этот перечень необходимо поддерживать в рабочем состоянии в файле менеджмента риска.
Также должны быть рассмотрены и зарегистрированы прогнозируемые последовательности событий, которые могут привести к созданию опасной ситуации.
Примечания
1 В качестве памятной записки могут быть использованы примеры возможных опасностей, перечисленные в приложении D и разделе В.2 для изделий для in vitro диагностики.
2 Для идентификации неизвестных ранее опасностей можно использовать методы систематизации, применяемые в конкретной ситуации (см. приложение F).
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
4.4 Определение риска (рисков) для каждой опасности (этап 3)
Риск(и) для каждой опасности, как в условиях нормальной эксплуатации, так и для аварийной ситуации, необходимо определять с помощью доступной информации или данных. В случае опасностей, для которых не может быть определена вероятность причинения вреда, должен быть составлен перечень возможных последствий таких опасностей. Определение риска(ов) должно быть зарегистрировано в файле менеджмента риска.
Любая система, используемая для качественной или количественной градации определения уровней вероятности или тяжести, должна быть зарегистрирована в файле менеджмента риска.
Примечания
1 Определение риска включает в себя анализ вероятности его возникновения и тяжести последствий. В зависимости от области применения, возможно, достаточно рассмотреть лишь некоторые элементы процесса определения риска. Например, в некоторых конкретных случаях нет необходимости продолжать определение риска после первичного анализа опасности и ее последствий.
2 Определение риска может быть количественным или качественным. Методы определения риска, включая методы, являющиеся следствием систематических неисправностей, представлены в приложении Е настоящего стандарта. Раздел В.3 настоящего стандарта содержит информацию, полезную при определении риска для медицинских изделий для in vitro диагностики.
3 Некоторые возможные методы анализа риска представлены в приложении F настоящего стандарта.
4 Информацию или данные для определения риска можно получить, например, из следующих источников:
- опубликованные стандарты;
- научно-техническая информация;
- данные по эксплуатации уже применяемых аналогичных медицинских изделий, включая опубликованные сведения об инцидентах;
- данные испытаний на эксплуатационную пригодность типичными пользователями;
- клинические данные;
- результаты исследований;
- экспертные оценки;
- схемы внешней оценки качества.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
5 Оценивание риска (этап 4)
Для каждой идентифицированной опасности изготовитель должен, применяя критерии плана менеджмента риска, решить, является ли оцениваемый риск(и) настолько низким(и), что нет смысла в его (их) дальнейшем уменьшении. При положительном ответе на этот вопрос требования 6.2-6.6 для конкретной опасности не применяют (т.е. после выполнения требований 6.1 выполняют требования 6.7). Результаты оценивания риска(ов) регистрируют в файле менеджмента риска.
Примечания
1 Руководство по принятию решения о допустимости риска приведено в Е.3, приложение Е.
2 Применение необходимых стандартов для разработки критериев проектирования медицинских изделий может являться частью деятельности по управлению риском, что обуславливает применение требований 6.3-6.6.
Соответствие настоящему разделу проверяют контролем файла менеджмента риска.
6.1 Уменьшение риска
При необходимости уменьшения риска(ов) изготовитель должен следовать описанному в 6.2-6.7 процессу управления риском(ами) так, чтобы связанный с каждой опасностью остаточный риск(и) был(и) квалифицирован(ы) как допустимый(е).
6.2 Анализ возможностей (этап 5)
Изготовитель должен идентифицировать меры по управлению риском(ами), необходимые для уменьшения риска(ов) до приемлемого уровня. Управление риском(ами) должно представлять собой интегрированный подход, при котором изготовитель должен применять в порядке приоритета один из нижеперечисленных:
a) безопасность, обеспечиваемая проектом и конструкцией медицинского изделия;
b) средства защиты или защитные меры, предусмотренные в процессе производства медицинского изделия;
c) информация по безопасности.
Примечания
1 Меры по управлению риском могут уменьшить тяжесть возможного вреда или вероятность причинения вреда, или и то и другое вместе.
2 Стандарты, устанавливающие общие технические требования, рассматривают безопасность, обеспечиваемую проектом и конструкцией, защитные меры и безопасность, обеспечиваемую соответствующей информацией, для многих медицинских изделий. Это следует учитывать в процессе менеджмента риска. См. также приложение G настоящего стандарта.
Выбранные меры по управлению риском необходимо зарегистрировать в файле менеджмента риска.
Если в процессе анализа возможностей изготовитель определяет, что дальнейшее уменьшение риска не имеет практического значения, он должен выполнить анализ соотношения риск/польза для остаточного риска (см. 6.5); в противном случае изготовитель должен продолжать выполнение выбранных мер по управлению риском.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
6.3 Выполнение мер по управлению риском (этап 6)
Изготовитель должен выполнять меры по управлению риском, выбранные в 6.2. Меры по управлению риском должны быть зарегистрированы в файле менеджмента риска.
Результативность мер по управлению риском должна быть верифицирована, а результаты верификации зарегистрированы в файле менеджмента риска.
Выполнение мер по управлению риском должно быть верифицировано. Результаты этой верификации также должны быть зарегистрированы в файле менеджмента риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
6.4 Оценивание остаточного риска (этап 7)
Любой остаточный риск, сохраняющийся после выполнения мер по управлению риском, необходимо оценивать по критериям, установленным в плане менеджмента риска. Результаты такого оценивания должны быть зарегистрированы в файле менеджмента риска.
Если остаточный риск не удовлетворяет установленным критериям, следует применить дополнительные меры по управлению риском (см. 6.2).
Если остаточный риск оценивают как допустимый, вся соответствующая информация по остаточному риску должна быть включена в соответствующие эксплуатационные документы, прилагаемые изготовителем.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска и эксплуатационных документов.
6.5 Анализ соотношения риск/польза (этап 8)
Если остаточный риск по критериям, установленным в плане менеджмента риска, был квалифицирован как недопустимый, а дальнейшие меры по управлению риском сочтены практически нецелесообразными, изготовитель должен собрать и проанализировать данные и литературу о медицинской пользе с точки зрения предусмотренного применения/предусмотренного назначения, чтобы определить, превышает ли она остаточный риск. Если собранные доказательства свидетельствуют о том, что медицинская польза не превышает остаточный риск, риск считается недопустимым. Если медицинская польза превышает остаточный риск, можно перейти к выполнению требований 6.6. Информация, необходимая для объяснения решения в отношении остаточного риска, должна быть включена в эксплуатационные документы, предоставляемые изготовителем. Результаты такого оценивания должны быть зарегистрированы в файле менеджмента риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска и эксплуатационных документов.
6.6 Другие возможные опасности (этап 9)
Меры по управлению риском необходимо анализировать, чтобы идентифицировать возможность создания ими других опасностей. Если какие-либо меры по управлению риском могут порождать любые новые опасности, то необходимо оценивать связанный с ними риск(и) (см. 4.4). Результаты такого анализа должны быть зарегистрированы в файле менеджмента риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
6.7 Полнота оценивания риска (этап 10)
Изготовитель должен обеспечивать оценивание риска(ов) всех идентифицированных опасностей. Результаты такого оценивания должны быть зарегистрированы в файле менеджмента риска.
Соответствие настоящему пункту проверяют контролем файла менеджмента риска.
7 Оценивание полного остаточного риска (этап 11)
После выполнения и верификации всех мер по управлению риском изготовитель должен принять решение о допустимости полного остаточного риска, создаваемого медицинским изделием, с точки зрения критериев, установленных в плане менеджмента риска. Если полный остаточный риск был квалифицирован как недопустимый с точки зрения критериев, установленных в плане менеджмента риска, изготовитель должен собрать и проанализировать данные и литературу о медицинской пользе с точки зрения предусмотренного применения/предусмотренного назначения изделия, чтобы установить, превышает ли медицинская польза полный остаточный риск. Если собранные доказательства свидетельствуют о том, что медицинская польза не превышает полный остаточный риск, он считается недопустимым. Результаты оценивания полного остаточного риска должны быть зарегистрированы в файле менеджмента риска.
Соответствие настоящему разделу проверяют контролем файла менеджмента риска.
8 Отчет по менеджменту риска (этап 12)
Результаты процесса менеджмента риска должны быть зарегистрированы в отчете по менеджменту риска. Отчет по менеджменту риска должен обеспечивать возможность осуществления прослеживаемости для каждой опасности при анализе риска, оценивании риска, выполнении и верификации мер по управлению риском, а также содержать оценку допустимости остаточного риска(ов). Отчет по менеджменту риска должен быть частью файла менеджмента риска.
Примечание - Отчет может быть на бумажном или электронном носителе.
Соответствие настоящему разделу проверяют контролем отчета по менеджменту риска.
9 Постпроизводственная информация (этап 13)
Изготовитель должен установить и поддерживать в рабочем состоянии систематическую процедуру анализа информации, собранной о медицинском изделии или аналогичных изделиях на постпроизводственном этапе. Эта информация должна быть оценена с точки зрения достижения надлежащей безопасности, в особенности в следующих вопросах:
a) существуют ли не опознанные ранее опасности;
b) не стал ли недопустимым риск(и), проистекающий(ие) из какой-либо опасности;
c) не стала ли первоначальная оценка недействительной по другим причинам?
При положительном ответе на любой из вышеуказанных вопросов результаты оценивания необходимо вернуть на вход процесса менеджмента риска (см. 4.4).
Отчет о соответствующих этапах процесса менеджмента риска для медицинского изделия необходимо рассматривать в свете полученной информации по безопасности. При наличии возможности изменения остаточного риска(ов) или его (их) допустимости следует оценить влияние такого изменения на ранее выполненные меры по управлению риском.
Результаты такого оценивания должны быть зарегистрированы в файле менеджмента риска.
Примечания
1 Отдельные аспекты постпроизводственного мониторинга рассматриваются в национальных или региональных регламентах. В некоторых случаях могут быть задействованы дополнительные меры, например перспективное постпроизводственное оценивание.
2 См. также 4.14 [3].
3 Информация может быть получена на любом этапе жизненного цикла медицинского изделия - от начального до постпроизводственного этапа включительно.
Соответствие настоящему разделу проверяют контролем документации процесса менеджмента риска и файла менеджмента риска.
Приложение А
(справочное)
Вопросы, на которые следует ответить при определении характеристик, влияющих на безопасность медицинского изделия
А.1 Общие положения
Первым этапом идентификации опасностей является анализ характеристик медицинского изделия, влияющих на безопасность его применения. Один из способов выполнения данного этапа - составление перечня вопросов, которые могут возникнуть при изготовлении, применении и конечной утилизации медицинского изделия. Если задавать эти вопросы от имени всех заинтересованных лиц (например пользователей, специалистов по уходу, пациентов и т.д.), можно получить более полную картину о возможных опасностях. Ответы на приведенные ниже вопросы могут помочь в идентификации возможных опасностей при работе с анализируемым медицинским изделием.
Этот перечень не является исчерпывающим, но следует быть осторожным при добавлении вопросов, которые могут быть отнесены к конкретному медицинскому изделию.
А.2 Вопросы
А.2.1 Каково предусмотренное применение/предусмотренное назначение и как следует применять медицинское изделие?
Рассматривают:
- предполагаемого пользователя, его умственные и физические возможности, квалификацию и подготовку;
- эргономические аспекты, условия эксплуатации, кто будет устанавливать изделие и сможет ли пациент сам управлять эксплуатацией медицинского изделия или влиять на нее.
Особое внимание рекомендуется уделять предполагаемым пользователям со специфическими потребностями, например лицам с физическими недостатками, пожилым людям, детям. Этим лицам может понадобиться помощь другого лица в эксплуатации медицинского изделия.
Предусмотрена ли эксплуатация медицинского изделия лицами с различным уровнем подготовки и различной общей культурой?
Какую роль отводят медицинскому изделию в диагностике, профилактике, мониторинге, лечении или облегчении болезни, компенсации травм или физических недостатков, замещении или модификации частей тела или управлении зачатием?
Предназначено ли медицинское изделие для жизнеобеспечения или для поддержания жизни?
Необходимо ли вмешательство специалистов при отказе медицинского изделия?
Необходимо ли уделять особое внимание проектным характеристикам интерфейса, которые могут привести к непроизвольным ошибкам эксплуатации изделия (см. А.2.27)?
А.2.2 Предполагается ли контакт медицинского изделия с пациентом или другими лицами?
Рассматривают: характер предполагаемого контакта, т.е. поверхностный контакт, инвазивный контакт и/или имплантация и, в каждом случае, длительность и частоту контакта.
А.2.3 Какие материалы и/или компоненты входят в состав медицинского изделия или использованы совместно либо в контакте с ним?
Рассматривают характеристики, влияющие на безопасность изделия.
А.2.4 Может ли энергия быть передана пациенту и/или выработана пациентом?
Рассматривают: вид передаваемой энергии, управление ею, ее качество, количество и длительность воздействия.
А.2.5 Вводят ли пациенту и/или выводят из него какие-либо вещества?
Рассматривают: вводимые пациенту или выводимые из него вещества; данные о том, одно это вещество или группа веществ; сведения о максимальной и минимальной скорости введения (выведения) вещества и управлении этим процессом.
А.2.6 Проводят ли в медицинском изделии обработку биологических веществ для их последующего использования?
Рассматривают тип обработки и обрабатываемое вещество(а) (например аутотрансфузия, диализ).
А.2.7 Стерильно ли поставляемое медицинское изделие или оно предназначено для стерилизации пользователем, или применяют другие виды микробиологической обработки?
Рассматривают: сведения о том, предназначено ли медицинское изделие для одноразового или многоразового применения; данные об упаковке, сроках хранения, любых ограничениях числа повторных применений или предполагаемых видах стерилизации.
А.2.8 Предназначено ли медицинское изделие для рутинных очистки и дезинфекции, выполняемых пользователем?
Рассматривают виды применяемых чистящих и дезинфицирующих средств и любые ограничения числа циклов очистки. Кроме того, на эффективность рутинной очистки и дезинфекции может влиять конструкция медицинского изделия.
А.2.9 Предназначено ли медицинское изделие для изменения среды, окружающей пациента?
Рассматривают: температуру, влажность, состав газов воздуха, давление и освещенность.
А.2.10 Могут ли быть проведены измерения?
Рассматривают: измеряемые переменные, правильность и точность результатов измерения.
А.2.11 Является ли медицинское изделие интерпретирующим?
Рассматривают: сведения о том, выдает ли медицинское изделие заключения на основе входных или накопленных данных; информацию о применяемых алгоритмах и доверительных пределах.
А.2.12 Предусмотрено ли применение медицинского изделия в сочетании с медикаментами или другими медицинскими средствами?
Рассматривают: идентификацию любых применяемых медикаментов или других медицинских средств, возможных проблем, связанных с их взаимодействием; реакцию пациента на получаемое лечение.
А.2.13 Могут ли произойти нежелательные выделения энергии или веществ?
Рассматривают в связи с возможным выделением энергии: шум и вибрацию, теплоту, излучение (включая ионизирующее, неионизирующее и ультрафиолетовое/видимое/инфракрасное излучения), температуру на контактных поверхностях, токи утечки, электрические и/или магнитные поля.
Рассматривают в связи с возможным выделением веществ: выведение химических веществ, продуктов жизнедеятельности и воды, содержащейся в организме.
А.2.14 Чувствительно ли медицинское изделие к воздействию окружающей среды?
Рассматривают: производственную среду, транспортные средства, условия хранения. К ним относят освещенность, температуру, вибрации, утечки, чувствительность к изменениям энергоснабжения и охлаждения, электромагнитные помехи.
А.2.15 Воздействует ли медицинское изделие на окружающую среду?
Рассматривают: воздействие средств энергоснабжения и охлаждения, выделение токсичных веществ, генерирование электромагнитных помех.
А.2.16 Имеются ли необходимые расходные материалы или принадлежности, связанные с медицинским изделием?
Рассматривают спецификации на расходные материалы или принадлежности, связанные с медицинским изделием, и любые ограничения в их выборе для пользователя.
А.2.17 Необходимы ли техническое обслуживание и/или калибровка?
Рассматривают: сведения о том, должны ли техническое обслуживание и/или калибровка быть выполнены оператором, пользователем или специалистом; необходимы ли специальные вещества или оборудование для надлежащего технического обслуживания и/или калибровки.
А.2.18 Входит ли в состав медицинского изделия программное обеспечение?
Рассматривают сведения о том, должно ли программное обеспечение быть установлено, верифицировано, модифицировано или заменено пользователем и/или оператором.
А.2.19 Имеет ли медицинское изделие ограниченный срок хранения?
Рассматривают соответствующие маркировку или индикацию, утилизацию медицинского изделия.
А.2.20 Существуют ли отсроченные и/или длительные последствия эксплуатации?
Рассматривают эргономические и кумулятивные эффекты.
А.2.21 Какие механические силы могут воздействовать на медицинское изделие?
Рассматривают данные о том, управляет ли механическими силами, которые могут воздействовать на медицинское изделие, только пользователь или пользователь совместно с другими лицами.
А.2.22 Что определяет срок службы медицинского изделия?
Рассматривают старение изделия, истощение элементов питания.
А.2.23 Предназначено ли медицинское изделие для одноразового применения?
А.2.24 Необходимы ли безопасный демонтаж или утилизация медицинского изделия?
Рассматривают проблему отходов, возникающих при утилизации медицинского изделия. Например, следует ответить, содержит ли изделие токсичные или опасные вещества либо вещества, пригодные для переработки.
А.2.25 Необходимо ли специальное обучение пользователя для монтажа или эксплуатации медицинского изделия?
Рассматривают установку (монтаж) и передачу изделия пользователю, вероятность/возможность установки (монтажа) лицами без специальной подготовки.
А.2.26 Будут ли необходимы разработка или внедрение новых производственных процессов?
Внедрение новых производственных процессов на предприятиях-изготовителях следует рассматривать как возможный источник новых опасностей (опасности) (например внедрение новой технологии, нового уровня развития производства).
А.2.27 Является ли успешное применение медицинского изделия критически зависимым от человеческого фактора, такого как интерфейс пользователя?
Рассматривают особенности конструкции интерфейса пользователя, которые могут привести к ошибкам эксплуатации. Медицинское изделие следует проектировать так, чтобы оно не могло быть неправильно применено пользователем в условиях действия отвлекающих факторов, например, при управлении изделием, что обеспечивается применением символов, разработкой эргономических характеристик, проекта конструкции и схемы расположения элементов, иерархии действий, меню для изделий с микропроцессорным управлением, наглядностью предупреждений, слышимостью сигналов тревоги, стандартной цветовой кодировкой. С этой целью следует также ответить на приведенные ниже вопросы, но не ограничиваться ими.
А.2.27.1 Имеет ли медицинское изделие соединительные части или принадлежности?
Рассматривают: наличие возможности неправильных соединений, отличие от соединений в других изделиях и сходство с ними, усилия, необходимые для соединения, обратную связь при повреждении соединения, а также чрезмерно или недостаточно туго затянутое соединение.
А.2.27.2 Имеет ли медицинское изделие управляющий интерфейс?
Рассматривают: пространственное распределение, кодирование, группировку, схему пространственного распределения, режимы обратной связи, грубые ошибки, незначительные промахи, дифференциацию управления, видимость изображений, направление активирования или изменения, дискретность или непрерывность функционирования элементов управления, обратимость уставок или действий.
А.2.27.3 Имеет ли медицинское изделие функцию отображения информации?
Рассматривают: видимость изображения в различных условиях, ориентацию, заполнение и перспективы, четкость представления информации, единицы отображения информации, цветовое кодирование и доступность критической информации.
А.2.27.4 Имеет ли медицинское изделие меню управления?
Рассматривают: сложность и число уровней, осведомленность о состоянии, расположение уставок, способ передвижения между объектами, число этапов в одном действии, проблемы четкой последовательности действий и запоминания, соотношение важности и доступности функции управления.
А.2.28 Является ли медицинское изделие мобильным или портативным?
Рассматривают: зажимы, рукоятки, колеса, тормоза, механическую устойчивость и прочность.
Приложение В
(справочное)
Руководство по анализу риска в отношении изделий для in vitro диагностики
В.1 Общие положения
Настоящее приложение содержит дополнительные руководящие указания по анализу риска в отношении изделий для in vitro диагностики с учетом особенностей и специфических аспектов этих изделий. Применение изделий для in vitro диагностики не создает прямого риска для пациента или обследуемого лица, так как их не используют внутри или на теле человека. Однако при идентификации опасностей, связанных с изделиями для in vitro диагностики, возможны косвенные риски, приводящие к ошибочным решениям или способствующие их принятию. Кроме того, рекомендуется рассматривать опасности, связанные с применением изделий, и возникающие при этом риски.
В.2 Идентификация опасностей
В дополнение к опасностям, изложенным в приложении D, при идентификации возможных для пациента или обследуемого лица опасностей рекомендуется рассматривать:
- неоднородность внутри одной партии, расхождения между разными партиями;
- общеизвестные источники опасностей;
- эффекты износа;
- погрешности при идентификации проб;
- проблемы стабильности (при хранении, транспортировании, эксплуатации, после первоначального вскрытия контейнера);
- проблемы, связанные с отбором, приготовлением и стабильностью проб;
- неадекватные спецификации исходных данных;
- неадекватные характеристики испытаний.
Возможную опасность для пользователя могут представлять радиоактивные, инфекционные, токсичные или другие опасные ингредиенты реактивов, а также конструкция упаковки. Применительно к инструментам следует рассматривать проблему возможного загрязнения при уходе, работе с ними и техническом обслуживании в дополнение к неспецифическим опасностям (например энергетическим), связанным с использованием инструментов.
В.3 Определение риска
При определении риска для каждой опасности рекомендуется учитывать:
- степень надежности аналитических результатов (медицинское заключение по результатам аналитических исследований);
- результаты проверки достоверности;
- наличие и применение элементов управления;
- средства измерения/методики обеспечения качества, применяемые в медицинских лабораториях;
- возможность выявления дефектов (погрешностей);
- ситуации применения (например при оказании скорой помощи);
- профессиональное /непрофессиональное применение;
- способ представления информации.
Приложение С
(справочное)
Руководство по процедуре анализа риска в отношении токсикологических опасностей
C.1 Общие положения
Настоящее приложение содержит руководящие указания по применению анализа риска к токсикологическим опасностям. Токсикологические опасности являются результатом воздействия химических веществ, наносящих биологический вред. В [5] установлены общие принципы биологического оценивания материалов/медицинских изделий.
Необходимо прилагать все усилия, чтобы избежать проведения необязательных испытаний на животных. Следует обратить внимание на [6], содержащий требования к защите животных, и на соответствующие национальные и региональные регламенты, предписывающие не проводить испытания на животных при возможности научного обоснования такого решения.
С.2 Определение рисков, связанных с токсикологическими опасностями
С.2.1 Необходимые для рассмотрения факторы
При анализе рисков, связанных с токсикологическими опасностями, следует учитывать:
- химические свойства материалов;
- опыт предыдущего применения материалов;
- данные испытаний на биологическую безопасность.
Количество необходимых данных и глубина исследования могут быть изменены в зависимости от предусмотренного применения/предусмотренного назначения изделия, а также от характера и продолжительности контакта с пациентом. Требования к данным обычно менее строги для упаковочных материалов, медицинских изделий, контактирующих с неповрежденной кожей, и для любых компонентов медицинских изделий, не вступающих в непосредственный контакт с тканями организма, слизистыми оболочками или поврежденной кожей, физиологическими жидкостями.
Для определения потребности в дополнительных данных следует проанализировать современные знания в области материалов/медицинских изделий, описанные в научной литературе, предшествующую клиническую практику, а также другие данные. В некоторых случаях может возникнуть необходимость получить данные о химическом составе, остаточных продуктах (например при процессах стерилизации), данных биологических испытаний.
С.2.2 Химические свойства материалов
Информация, характеризующая химическую идентичность и биологическую реакцию материалов, полезна при оценке предусмотренного применения/предусмотренного назначения медицинского изделия. К факторам, способным повлиять на биологическую совместимость материала, относят:
- идентичность, концентрацию, доступность и токсичность всех составляющих (например добавок, средств обработки, мономеров, катализаторов, продуктов реакции);
- влияние биологического распада и коррозии на материалы.
Если при производстве, обработке, хранении или деградации материала могут быть применены или образованы реагирующие или опасные ингредиенты, рекомендуется рассмотреть возможное воздействие остаточных продуктов. При этом может возникнуть необходимость в информации о концентрации и/или выщелачивании остаточных продуктов. Это могут быть экспериментальные данные или информация о химических свойствах применяемых материалов. Если необходимые данные (например данные о полном химическом составе) недоступны изготовителю по причине конфиденциальности, рекомендуется верифицировать определение соответствия применяемого материала в предполагаемой ситуации.
С.2.3 Предыдущий опыт применения
Рекомендуется проанализировать всю имеющуюся информацию о предыдущем опыте применения каждого материала или предполагаемой добавки и о возникших побочных реакциях. Однако предыдущий опыт применения ингредиента или материала не всегда можно использовать в аналогичных случаях. Следует учитывать предусмотренное применение/предусмотренное назначение, концентрацию ингредиентов и текущую токсикологическую информацию.
С.2.4 Данные испытаний на биологическую безопасность
Руководящие указания о том, какие испытания предназначены для каждого конкретного случая применения, содержался в [5]. Необходимость испытаний следует рассматривать для каждого случая отдельно в свете имеющихся данных, чтобы избежать проведения необязательных испытаний.
Приложение D
(справочное)
Примеры возможных опасностей и сопутствующих факторов, связанных с медицинскими изделиями
D.1 Общие положения
Настоящее приложение содержит перечень возможных опасностей и сопутствующих факторов, связанных с медицинскими изделиями. Этот перечень не является исчерпывающим, однако может помочь идентифицировать опасности, связанные с конкретным медицинским изделием.
D.2 Энергетические опасности и сопутствующие факторы
К энергетическим опасностям и сопутствующим факторам относят:
- электричество;
- теплоту;
- механическую силу;
- ионизирующую радиацию;
- неионизирующую радиацию;
- движущиеся части;
- непредусмотренное движение;
- взвешенные вещества;
- отказ изделия, поддерживающего жизнь пациента;
- давление (например при разрыве сосудов);
- акустическое давление;
- вибрацию;
- магнитные поля (например ядерно-магнитный резонанс).
D.3 Биологические опасности и сопутствующие факторы
К биологическим опасностям и сопутствующим факторам относят:
- биологическое загрязнение;
- биологическую несовместимость;
- неадекватный химический состав;
- токсичность;
- аллергенность;
- мутагенность;
- онкогенность;
- тератогенность;
- канцерогенность;
- повторную и/или перекрестную инфекцию;
- пирогенность;
- неспособность поддерживать гигиеническую безопасность;
- вырождение.
D.4 Опасности, связанные с окружающей средой, и сопутствующие факторы
К опасностям, связанным с окружающей средой, и сопутствующим факторам относят:
- электромагнитные поля;
- восприимчивость к электромагнитной интерференции;
- эмиссию электромагнитной интерференции;
- неадекватное электропитание;
- неадекватную подачу хладагентов;
- хранение или работу в непредусмотренных производственных условиях;
- несовместимость с другими изделиями, с которыми предусмотрено применение;
- случайное механическое повреждение;
- загрязнение побочными продуктами применения и/или утилизации медицинского изделия.
D.5 Опасности, возникающие вследствие неадекватного выхода энергии и выделения веществ
К опасностям, возникающим вследствие неадекватного выхода энергии и выделения веществ, относят:
- электричество;
- радиацию;
- объем;
- давление;
- подачу медицинских газов;
- подачу анестетиков.
D.6 Опасности, связанные с применением медицинского изделия, и сопутствующие факторы
К опасностям, связанным с применением медицинского изделия, и сопутствующим факторам относят:
- неадекватную маркировку;
- неадекватные инструкции по эксплуатации, например:
1) неадекватную спецификацию принадлежностей, применяемых с медицинским изделием,
2) неадекватное описание предварительных проверок,
3) чрезмерную сложность инструкций по эксплуатации,
4) неадекватное описание оказываемых услуг и технического обслуживания;
- эксплуатацию изделия неквалифицированным/необученным персоналом;
- обоснованно прогнозируемое неправильное применение;
- недостаточное предупреждение о побочных эффектах;
- неадекватное предупреждение об опасностях повторного применения одноразового медицинского изделия;
- некорректное измерение и другие метрологические аспекты (например использование нестандартизованных единиц измерения, неаттестованных приборов);
- несовместимость с расходными материалами/принадлежностями/другими медицинскими изделиями;
- острые края или наконечники.
D.7 Несоответствующий, неадекватный или чрезмерно сложный интерфейс пользователя (обмен информацией между человеком и машиной)
К несоответствующему, неадекватному или чрезмерно сложному интерфейсу пользователя относят:
- ошибки и заблуждения;
- упущения и ошибки при когнитивном воспроизведении;
- промахи и просчеты (умственные и физические);
- нарушение или сокращение инструкций, процедур;
- сложную или запутанную систему управления;
- неоднозначное или неясное состояние изделия;
- неоднозначное или неясное представление уставок, измерений или другой информации;
- неправильное представление результатов;
- недостаточные видимость, слышимость или тактильность;
- ненадлежащее расположение элементов управления действиями или представление информации о реальном состоянии;
- режимы или расположение элементов, противоречащие имеющемуся оборудованию.
D.8 Опасности, возникающие при функциональном отказе, в процессе технического обслуживания, при старении, и сопутствующие факторы
К опасностям, возникающим при функциональном отказе, в процессе технического обслуживания, при старении, и сопутствующим факторам относят:
- ошибки при передаче данных;
- отсутствие или неадекватность инструкций по техническому обслуживанию, включая неадекватное описание функциональных проверок после технического обслуживания;
- неадекватное техническое обслуживание;
- отсутствие адекватного определения окончания срока эксплуатации медицинского изделия;
- потерю диэлектрической/механической целостности;
- неадекватную упаковку (загрязнение и/или порчу медицинского изделия);
- многократное применение и/или неправомерное повторное применение;
- ухудшение функциональных свойств (например постепенное сужение магистралей для жидкости/газа или изменение сопротивления потоку, электрической проводимости) в результате многократного применения.
Приложение Е
(справочное)
Концепции риска, применимые к медицинским изделиям
Е.1 Определение риска
Для определения риска можно использовать различные методы. Настоящий стандарт не требует применения какого-либо конкретного метода, но требует определения риска (см. 4.4). Количественное определение риска возможно при наличии необходимых данных. Методы количественного определения риска могут просто заключаться в адаптации качественного метода или применении какого-либо альтернативного подхода.
Пример схематического представления риска, показанный на рисунке Е.1, может быть использован как часть качественного метода определения риска. Рисунок Е.1 включен в стандарт только в качестве иллюстрации метода. Это не означает, что такое схематическое представление применяют для медицинских изделий повсеместно. Если для определения риска используют метод схематического представления риска, то рекомендуется обосновать применение схематического представления конкретного риска и привести его интерпретацию.

Рисунок Е.1 - Схематическое представление трех областей риска
Концепция риска является комбинацией двух следующих компонентов:
- вероятности причинения вреда, то есть возможной частоты его причинения;
- последствий причиненного вреда, то есть его тяжести.
При определении риска рекомендуется рассматривать инициирующие происшествия или обстоятельства, последовательность нежелательных событий, любые смягчающие факторы, а также характер и частоту возможных вредных последствий идентифицированных опасностей. Риск следует определять в терминах, облегчающих принятие решений по управлению риском. При анализе риска рекомендуется его компоненты, т.е. вероятность и тяжесть, рассматривать раздельно.
Е.2 Вероятность риска
Е.2.1 Уровни вероятности риска
В конкретных обстоятельствах, при наличии достаточного количества данных предпочтительным является разделение уровней вероятности риска по количественным характеристикам. Если это невозможно, изготовитель должен привести описание по качественным признакам. При разделении уровней вероятности риска по качественным признакам изготовитель может использовать дескрипторы, предназначенные для конкретного медицинского изделия. Фактически это понятие представляет собой континуум, однако на практике можно применять конкретное число уровней. В этом случае изготовитель устанавливает необходимое число уровней и способ их определения. Уровни вероятности риска могут быть выражены описательными определениями (например "невероятная", "маловероятная", "незначительная", "случайная", "вероятная", "частая") или символами (Р1, Р2 и т.д.).
При определении уровней вероятности рассматривают инициирующие события и обстоятельства, а также последовательность нежелательных событий. При этом отвечают на вопрос, возникает ли опасность:
- при отсутствии отказа изделия;
- в случае отказа;
- только при условии многократных отказов.
Уровень вероятности каждого происходящего нежелательного события определяют на стадии идентификации опасности. Для определения уровня вероятности риска обычно применяют следующие способы:
- использование соответствующих данных предыдущего опыта применения;
- прогнозирование вероятности риска с помощью аналитических и моделирующих методов;
- использование экспертных оценок.
Все эти способы можно применять как по отдельности, так и совместно. Первые два способа дополняют друг друга. Там, где возможно, рекомендуется применять оба способа. В этом случае их можно использовать для независимых перекрестных проверок, что позволит повысить достоверность результатов. Если эти способы не могут быть использованы или являются недостаточными, может возникнуть необходимость в экспертных оценках.
Некоторые опасности возникают вследствие скорее систематических, чем случайных отказов (например опасности, вызванные отказами программного обеспечения, связаны с систематическими отказами). Систематические отказы рассмотрены в Е.4.3.
Е.2.2 Уровни тяжести риска
При разделении уровней тяжести риска по качественным признакам изготовитель может использовать дескрипторы, предназначенные для конкретного медицинского изделия. Фактически это понятие представляет собой континуум, однако на практике можно применять конкретное число уровней. В этом случае изготовитель устанавливает необходимое число уровней и способ их определения. Уровни тяжести риска могут быть выражены описательными определениями (например "пренебрежимо малая", "незначительная", "критическая", "серьезная", "катастрофическая") или символами (S1, S2 и т.д.).
Изготовитель должен применять разделение на уровни тяжести риска к конкретному медицинскому изделию с учетом как краткосрочных, так и долгосрочных эффектов.
Е.3 Допустимый риск
Е.3.1 Общие положения
Настоящий стандарт не устанавливает уровни допустимого риска. Методы определения допустимого риска включают в себя:
- применение стандартов, устанавливающих требования, соответствие которым должно свидетельствовать о достижении допустимого риска для медицинских изделий конкретных видов и конкретных рисков;
- исполнение соответствующих руководящих указаний, например тех, которые применяют в концепции единичного отказа (подробнее см. [7], 9.10);
- сравнение уровней допустимого риска, известных по уже применяемым медицинским изделиям.
Риск рекомендуется считать допустимым, только если он мал по сравнению с ожидаемой пользой.
Любой риск можно отнести к одной из трех областей:
1) широко допустимого риска;
2) минимального практически достижимого риска;
3) недопустимого риска.
Пример схематического изображения трех областей риска представлен на рисунке Е.1. Понятие трех областей риска должно быть применено к конкретному медицинскому изделию.
Примеры количественного определения вероятности и тяжести риска можно найти в некоторых стандартах, указанных в [7]-[9], [12]. Пользователям настоящего стандарта рекомендуется определять уровни вероятности и тяжести риска в отношении собственных случаев применения.
Е.3.2 Область широко допустимого риска
В некоторых случаях оцениваемый риск настолько мал по сравнению с другими рисками и с точки зрения пользы от применения медицинского изделия, что им можно пренебречь. В таких случаях риск является допустимым и нет необходимости в принятии активных мер по его управлению.
Е.3.3 Область минимального практически достижимого риска
Можно предположить, что любой риск, связанный с медицинским изделием, является допустимым при наличии улучшения прогнозов для пациента. Но такой подход служит оправданием для принятия необоснованного риска. Любой риск следует уменьшать до минимального практически достижимого уровня с учетом пользы от принятия риска, а также возможности и целесообразности его дальнейшего уменьшения.
К целесообразности относят способность изготовителя уменьшить риск. Целесообразность имеет две составляющие:
a) техническую;
b) экономическую.
К технической целесообразности относят способность изготовителя уменьшить риск без учета затрат. К экономической целесообразности относят способность изготовителя уменьшить риск без превращения материального обеспечения производства медицинского изделия в экономически необоснованный проект. Факторы стоимости и доступности рассматривают при решении вопроса о том, какие меры целесообразны в той степени, в какой они влияют на сохранение, поддержание или улучшение здоровья человека.
Обычно основные риски следует уменьшать даже при значительных материальных затратах. Если речь идет об области широко допустимого риска, можно считать достаточным достижение оптимального соотношения риск/польза.
Е.3.4 Область недопустимого риска
Некоторые риски при отсутствии возможности их уменьшения можно рассматривать как недопустимые в любом случае.
Е.3.5 Решения о допустимости риска
Необходимо четко разграничивать риски, которые так малы, что нет необходимости в их рассмотрении, и риски, которые больше первых, но допустимы ввиду ожидаемой пользы и нецелесообразности их уменьшения. После идентификации опасности и определения риска необходимо решить главный вопрос - является ли риск настолько низким, что его можно не рассматривать и, следовательно, не уменьшать. Это решение должно быть принято для каждой опасности.
Если на первом этапе принимают решение, что риск не является пренебрежимо малым, то следующим этапом будет работа по уменьшению риска. Уменьшение риска может быть целесообразным или нецелесообразным, но этот вопрос следует рассматривать. На втором этапе возможны следующие выводы:
- принятие одной или более мер по уменьшению риска может уменьшить риск до уровня, при котором не будет необходимости в его дальнейшем рассмотрении;
- независимо от того, возможно или нет некоторое уменьшение риска, его уменьшение до уровня, при котором нет необходимости в его рассмотрении, будет нецелесообразным.
В последнем случае риск рекомендуется уменьшить до минимального практически достижимого уровня, а затем сравнить возможный риск и ожидаемую пользу. Если польза превышает риск, то риск можно считать допустимым. Если польза не превышает риск, то риск считают недопустимым и от проекта следует отказаться.
Наконец, когда все риски признаны допустимыми, оценивают полный остаточный риск для того, чтобы убедиться в сохранении оптимального соотношения между возможным риском и ожидаемой пользой.
Таким образом, есть три проблемы, требующие принятия решения, когда возникают следующие вопросы о допустимости рисков:
1) является ли риск настолько низким, что нет необходимости в его рассмотрении?
2) есть ли еще причины для рассмотрения риска или же риск настолько мал, насколько это практически достижимо, и ожидаемая польза его перевешивает?
3) достигнуто ли общее оптимальное соотношение между всеми рисками и всей ожидаемой пользой?
Е.4 Причины отказов
Е.4.1 Виды отказов
Опасные ситуации могут быть следствием отказа системы. Существуют два возможных вида отказов:
1) случайные;
2) систематические.
Е.4.2 Случайный отказ
Статистическая вероятность отказа может быть определена для многих событий (например, вероятность отказа узла часто определяют исходя из вероятностей отказа его компонентов). В этом случае вероятности отказа может быть присвоено цифровое значение. Основным предположением является случайный характер отказов. Принято считать, что отказы аппаратного обеспечения могут носить случайный или систематический характер, а отказы программного обеспечения - систематический характер.
Е.4.3 Систематический отказ
Систематические отказы возникают при осуществлении любого вида деятельности и являются результатом ошибок (включая ошибки выполнения или невыполнения), когда определенные сочетания входных данных или окружающих условий могут привести к отказу.
Ошибки, приводящие к систематическим отказам, могут возникнуть как в аппаратном, так и в программном обеспечении в любой момент в процессе разработки, изготовления или технического обслуживания медицинского изделия. Примерами систематических отказов являются случаи, когда:
a) неверно рассчитанный плавкий предохранитель не позволяет предотвратить опасную ситуацию. Параметры плавкого предохранителя могли быть неверно определены, неправильно установлены при изготовлении или неправильно заменены при ремонте;
b) применение несоответствующего материала при замещении тазобедренного сустава приводит к чрезмерно быстрому износу и преждевременному отказу имплантата тазобедренного сустава. Примененный материал мог быть неверно специфицирован или неверно использован при изготовлении имплантата (например заказ несоответствующего материала у поставщика);
с) в базе данных программного обеспечения не предусмотрено состояние заполненной базы данных. Если база данных заполнена, неясно, что делать программному обеспечению. Возможным последствием может стать удаление существующих записей из базы данных с целью освобождения места для новых записей.
Точное определение частоты систематических отказов затруднительно. Это связано, в первую очередь, со следующими причинами:
a) измерение частоты систематических отказов требует больших затрат труда и денежных средств. Достижение приемлемого уровня достоверности результатов невозможно без измерения частоты отказов в течение длительного времени;
b) не достигнуто согласие по количественному методу определения частоты систематических отказов.
В тех случаях, когда для определения систематических отказов не может быть установлен соответствующий уровень достоверности, менеджмент риска рекомендуется осуществлять, исходя из тяжести вреда, причиняемого конкретной опасностью. Первоначальное определение риска возникновения систематических отказов следует основывать на предположении, что систематический отказ происходит с неприемлемой частотой.
Существует взаимосвязь между качеством применяемых процессов разработки и возможностью возникновения или невыявления систематического отказа. Часто целесообразно определить необходимое качество процесса разработки с учетом тяжести последствий систематических отказов и результативности внешних мер по управлению риском. Чем тяжелее последствия и меньше результативность внешних мер по управлению риском, тем выше должно быть качество процесса разработки.
Приложение F
(справочное)
Информация о методах анализа риска
F.1 Общие положения
Настоящее приложение содержит руководящие указания относительно некоторых существующих методов вероятностного анализа безопасности медицинского изделия, которые можно применять согласно 4.3. Эти методы являются дополнительными, и может возникнуть необходимость в использовании некоторых из них. Основной принцип заключается в поэтапном анализе возможных последствий предполагаемого события. Более детальные разъяснения приведены в [8].
F.2 Анализ характера и последствий отказов
Анализ характера и последствий отказов является, в первую очередь, качественным методом, с помощью которого систематически определяют и оценивают последствия отказа отдельного компонента медицинского изделия. Это индуктивный метод, использующий вопрос "Что случится на выходе, если ...?". Компоненты медицинского изделия анализируют последовательно, один за другим, что в основном соответствует условию единичного отказа. Анализ проводят в режиме "снизу-вверх", т.е. посредством перехода к следующему, более высокому, функциональному уровню системы.
Анализ характера и последствий отказов может быть расширен за счет исследования степени тяжести последствий, вероятности их возникновения и обнаружения соответственно, и может стать анализом характера, последствий и критичности отказов. Для выполнения такого анализа конструкция медицинского изделия должна быть известна во всех подробностях.
Анализ характера и последствий отказов может быть полезным методом рассмотрения ошибок человека. Такой анализ также можно применять для идентификации опасностей, предоставляя таким образом ценные входные данные для анализа "древа неисправностей" (см. F.3).
Недостатки этого метода связаны с трудностями, возникающими при наличии избыточной информации, рассмотрении и выполнении действий по ремонту и превентивному техническому обслуживанию, а также с ограниченными возможностями метода в условиях единичного отказа.
Более подробная информация по методам анализа характера и последствий отказов приведена в [9].
F.3 Анализ древа неисправностей
Анализ древа неисправностей является, в первую очередь, средством анализа опасностей, идентифицированных с помощью других методов; его начинают с положения о нежелательном последствии, называемом также "главным событием". Методом дедукции, начиная с главного события, идентифицируют возможные причины или характеры отказов, вызывающие нежелательные последствия, на более низком функциональном уровне системы. Следующая за этим поэтапная идентификация нежелательного функционирования системы с последовательным понижением уровней приводит к тому уровню системы, который обычно и представляет собой источник неисправности. Это позволяет установить последовательности, наиболее часто являющиеся причинами нежелательного события. Применение такого метода полезно при судебных разбирательствах.
Результаты представляют графически в виде древа неисправностей. На каждом уровне древа комбинации видов неисправностей описаны логическими операторами (И, ИЛИ и т.д.). Виды неисправностей, идентифицированные на древе, могут являться событиями, связанными с отказами аппаратного обеспечения, человеческим фактором или любыми другими происшествиями, ведущими к нежелательным последствиям. Виды неисправностей не ограничены условиями единичного отказа.
Анализ древа неисправностей позволяет систематически и в то же время достаточно гибко рассматривать различные факторы, включая человеческий. Анализ древа неисправностей используют, в первую очередь, при анализе риска в качестве инструмента определения вероятностей отказов. Графическое представление облегчает понимание поведения системы и входящих в нее факторов, но когда древо становится большим, его обработка может потребовать применения компьютерной системы. Эта особенность затрудняет верификацию древа неисправностей.
Более полная информация о процедурах анализа древа неисправностей приведена в [10].
F.4 Изучение опасностей и эксплуатационной пригодности
Изучение опасностей и эксплуатационной пригодности проводят аналогично анализу характера и последствий отказов на основании теоретической предпосылки, что нежелательные события обусловлены отклонениями от проекта или предусмотренной эксплуатации медицинского изделия. Изучение опасностей и эксплуатационной пригодности является систематическим методом. Этот метод был первоначально разработан для применения в химической промышленности. Поскольку применение данного метода в химической промышленности сосредоточено на отклонениях от проекта, существуют его альтернативные применения в области разработки медицинских изделий. Метод изучения опасностей и эксплуатационной пригодности можно применять к процессу функционирования конкретного медицинского изделия (например существующим методам/процессам диагностики, лечения или облегчения болезней, выступающим в качестве цели проекта) или к процессам, используемым при изготовлении или техническом обслуживании медицинского изделия (например стерилизации), которые могут оказывать значительное влияние на функционирование медицинского изделия. Для данного метода характерны две основные особенности:
1) использование группы специалистов, компетентных в области проектирования и применения конкретного медицинского изделия;
2) применение соответствующих ключевых слов для идентификации отклонений от нормальной эксплуатации.
Целями метода являются:
- составление полного описания медицинского изделия и его предусмотренного применения;
- систематический анализ каждой части предусмотренного применения/ предусмотренного назначения с целью определения причины отклонения от нормальных рабочих условий и предусмотренного проекта;
- определение последствий таких отклонений и принятие решения о том, могут ли эти последствия привести к возникновению опасностей или создать проблемы с эксплуатацией.
Последняя цель особенно важна применительно к процессам, используемым при изготовлении медицинских изделий, когда характеристики конкретного медицинского изделия зависят от процесса изготовления.
Приложение G
(справочное)
Другие стандарты, содержащие информацию об элементах менеджмента риска, описанных в настоящем стандарте
Таблица G.1 - Элементы менеджмента качества, связанные с элементами менеджмента риска
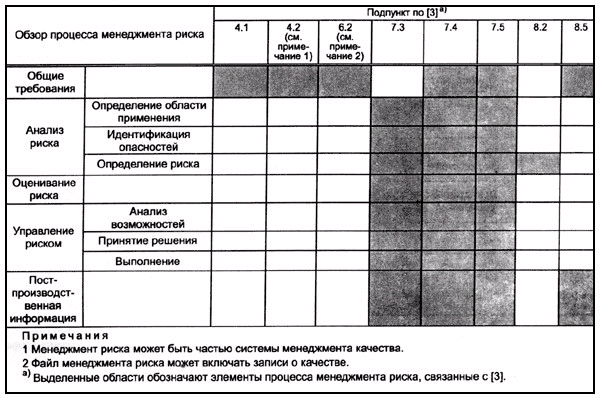
Таблица G.2 - Другие международные стандарты, связанные с элементами менеджмента риска

|
Приложение Н | |
|
Н.2 Обоснование раздела 2 Термины и определения Н.3 Обоснование раздела 3 Общие требования к менеджменту риска Н.3.2 Процесс менеджмента риска Н.3.3 Ответственность высшего руководства Н.3.4 Квалификация персонала Н.3.5 План менеджмента риска
В настоящем стандарте термин "файл менеджмента риска" применяют для обозначения намерения изготовителя разместить в конкретном месте или найти местоположение всех записей, относящихся к менеджменту риска. Это облегчает менеджмент риска и позволяет более эффективно провести аудит по настоящему стандарту.
Н.4.4 Определение риска(ов) для каждой опасности
Н.6.1 Уменьшение риска Н.6.2 Анализ возможностей Н.6.7 Полнота оценивания риска
Н.8 Обоснование раздела 8 Отчет по менеджменту риска
Н.9 Обоснование раздела 9 Постпроизводственная информация | |
Приложение J
(обязательное)
Сведения о соответствии национальных стандартов Российской Федерации ссылочным международным стандартам
Таблица J.1
|
Обозначение ссылочного международного стандарта |
Обозначение и наименование соответствующего национального стандарта |
|
МЭК 60601-1:1988 |
ГОСТ 30324.0-95 (МЭК 601-1-88)/ГОСТ Р 50267.09-92* (МЭК 601-1-88) Изделия медицинские электрические. Часть 1. Общие требования безопасности |
|
ИСО 13485:2003 |
|
|
ИСО 9000:2000 |
ГОСТ Р ИСО 9000-2001 Системы менеджмента качества. Основные положения и словарь |
|
ИСО 10993-1:2003 |
ГОСТ Р ИСО 10993.1-99 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования |
|
ИСО 14971-1:1998 |
ГОСТ Р ИСО 14971-1-99 Медицинские изделия. Управление риском. Часть 1. Применение анализа риска к медицинским изделиям |
________________
* Вероятно, ошибка оригинала. Следует читать ГОСТ 30324.0-95 (МЭК 601-1-88)/ГОСТ Р 50267.0-92 (МЭК 601-1-88). - Примечание .
Библиография
|
[1] |
МЭК 60601-1:1988 |
Изделия медицинские электрические. Часть 1. Общие требования безопасности |
|
|
(IEC 60601-1:1988) |
(Medical electrical equipment - Part 1: General requirements for safety) |
|
[2] |
ISO/IEC Guide 51:1999 |
Safety aspects - Guidelines for the inclusion in standards |
|
[3] |
ИСО 13485:2003 |
Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования |
|
|
(ISO 13485:2003) |
(Medical devices - Quality management systems - System requirements for regulatory purposes) |
|
[4] |
ИСО 9000:2000 |
Системы менеджмента качества. Основные положения и словарь |
|
(ISO 9000:2000) |
(Quality management systems - Fundamentals and vocabulary) | |
|
[5] |
ИСО 10993-1:2003 |
Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования |
|
|
(ISO 10993-1:2003) |
(Biological evaluation of medical devices - Part 1: Evaluation and testing) |
|
[6] |
ISO 10993-2:1992 |
Biological evaluation of medical devices - Part 2: Animal welfare requirements |
|
[7] |
IEC/TR 60513:1994 |
Fundamental aspects of safety standards for medical electrical equipment |
|
[8] |
IEC 60300-3-9:1995 |
Dependability management - Part 3: Application guide - Section 9: Risk analysis of technological systems |
|
[9] |
IEC 60812:1985 |
Analysis techniques for system reliability - Procedures for failure mode and effects analysis (FMEA) |
|
[10] |
IEC 61025:1990 |
Fault tree analysis (FTA) |
|
[11] |
Директива 93/42/ЕЭС от 14 июня 1993 г. |
О медицинских изделиях |
|
(Council Directive 93/42/EEC of 14 June 1993) |
(On medical devices) | |
|
[12] |
IEC 60601-1-4:2000 |
Medical electrical equipment - Part 1: General requirements for safety - 4: Collateral standard: Programmable electrical medical systems |
|
[13] |
ИСО 14971-1:1998 |
Медицинские изделия. Менеджмент риска. Часть 1. Применение анализа риска |
|
(ISO 14971-1:1998) |
(Medical devices - Risk management - Part 1: Application of risk analysis) |
Текст документа сверен по:
официальное издание
М.: Стандартинформ, 2006
Личный кабинет:
доступно после авторизации Разлив нефти: 12,6 тонн загрязненного песка убрали с пляжей в Анапе и...
Разлив нефти: 12,6 тонн загрязненного песка убрали с пляжей в Анапе и...  Создайте свой интернет-магазин на новой платформе ReadyScript
Создайте свой интернет-магазин на новой платформе ReadyScript  Хостинг, домены, VPS/VDS, размещение серверов
Хостинг, домены, VPS/VDS, размещение серверов
© 2007-2024 ООО «РуФокс»
о проекте
вакансии
хостинг
создание сайтов
реклама на сайте
наши партнеры
сообщить об ошибке